 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeExact Variance and Fano Factor for Arbitrary Level Crossings in Stationary Gaussian Processes
May 24, 2026Understanding the statistics of level crossings in stochastic processes is crucial across many scientific disciplines. The traditional Kac-Rice formula gives the mean rate of level crossings and has found broad use. However, that mean rate captures only a coarse summary of the crossing process. It depends entirely on local properties of the stochastic process at a given instant and is therefore blind to the correlation structure of the process over time. To understand whether crossing events, such as neuronal spikes, tend to cluster in time, spread apart, or exhibit more complex temporal organization, one must go beyond the mean rate and study higher-order crossing statistics. Here we go beyond the mean by deriving the exact analytical formulae for the variance and Fano factor of arbitrary level crossings in smooth stationary Gaussian processes. Our exact solution reveals how the full temporal correlation structure dictates whether crossings cluster or become regular. In systems with oscillatory correlations, such as a stochastic damped harmonic oscillator, a recent crossing suppresses an immediate subsequent one, producing sub-Poissonian statistics. However, as damping increases and oscillations disappear, a large and slow excursion above the threshold can produce multiple closely spaced crossings, yielding super-Poissonian statistics. In purely relaxational, non-oscillatory systems, such as a mean-reverting process driven by Ornstein-Uhlenbeck noise, the competition between the timescales of the driving noise and system relaxation produces a richer landscape, including reentrant transitions between sub- and super-Poissonian statistics as the threshold level is varied. Taken together, the exact variance and Fano factor derived here complement the Kac-Rice mean rate, enabling more robust parameter estimation and model selection across any setting where Gaussian processes are used.
MolCrystalFlow: Molecular Crystal Structure Prediction via Flow Matching
Feb 17, 2026Molecular crystal structure prediction represents a grand challenge in computational chemistry due to large sizes of constituent molecules and complex intra- and intermolecular interactions. While generative modeling has revolutionized structure discovery for molecules, inorganic solids, and metal-organic frameworks, extending such approaches to fully periodic molecular crystals is still elusive. Here, we present MolCrystalFlow, a flow-based generative model for molecular crystal structure prediction. The framework disentangles intramolecular complexity from intermolecular packing by embedding molecules as rigid bodies and jointly learning the lattice matrix, molecular orientations, and centroid positions. Centroids and orientations are represented on their native Riemannian manifolds, allowing geodesic flow construction and graph neural network operations that respects geometric symmetries. We benchmark our model against state-of-the-art generative models for large-size periodic crystals and rule-based structure generation methods on two open-source molecular crystal datasets. We demonstrate an integration of MolCrystalFlow model with universal machine learning potential to accelerate molecular crystal structure prediction, paving the way for data-driven generative discovery of molecular crystals.
Cross-View World Models
Feb 07, 2026World models enable agents to plan by imagining future states, but existing approaches operate from a single viewpoint, typically egocentric, even when other perspectives would make planning easier; navigation, for instance, benefits from a bird's-eye view. We introduce Cross-View World Models (XVWM), trained with a cross-view prediction objective: given a sequence of frames from one viewpoint, predict the future state from the same or a different viewpoint after an action is taken. Enforcing cross-view consistency acts as geometric regularization: because the input and output views may share little or no visual overlap, to predict across viewpoints, the model must learn view-invariant representations of the environment's 3D structure. We train on synchronized multi-view gameplay data from Aimlabs, an aim-training platform providing precisely aligned multi-camera recordings with high-frequency action labels. The resulting model gives agents parallel imagination streams across viewpoints, enabling planning in whichever frame of reference best suits the task while executing from the egocentric view. Our results show that multi-view consistency provides a strong learning signal for spatially grounded representations. Finally, predicting the consequences of one's actions from another viewpoint may offer a foundation for perspective-taking in multi-agent settings.
Open Materials Generation with Inference-Time Reinforcement Learning
Jan 31, 2026Continuous-time generative models for crystalline materials enable inverse materials design by learning to predict stable crystal structures, but incorporating explicit target properties into the generative process remains challenging. Policy-gradient reinforcement learning (RL) provides a principled mechanism for aligning generative models with downstream objectives but typically requires access to the score, which has prevented its application to flow-based models that learn only velocity fields. We introduce Open Materials Generation with Inference-time Reinforcement Learning (OMatG-IRL), a policy-gradient RL framework that operates directly on the learned velocity fields and eliminates the need for the explicit computation of the score. OMatG-IRL leverages stochastic perturbations of the underlying generation dynamics preserving the baseline performance of the pretrained generative model while enabling exploration and policy-gradient estimation at inference time. Using OMatG-IRL, we present the first application of RL to crystal structure prediction (CSP). Our method enables effective reinforcement of an energy-based objective while preserving diversity through composition conditioning, and it achieves performance competitive with score-based RL approaches. Finally, we show that OMatG-IRL can learn time-dependent velocity-annealing schedules, enabling accurate CSP with order-of-magnitude improvements in sampling efficiency and, correspondingly, reduction in generation time.
MolGuidance: Advanced Guidance Strategies for Conditional Molecular Generation with Flow Matching
Dec 13, 2025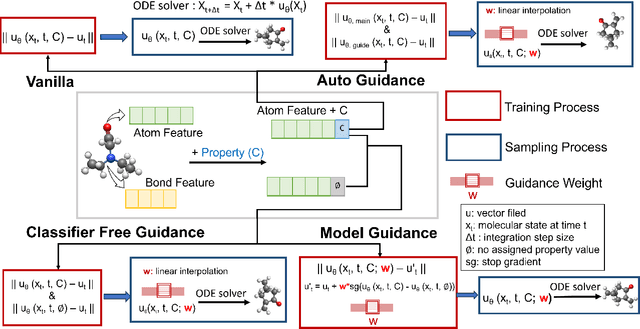

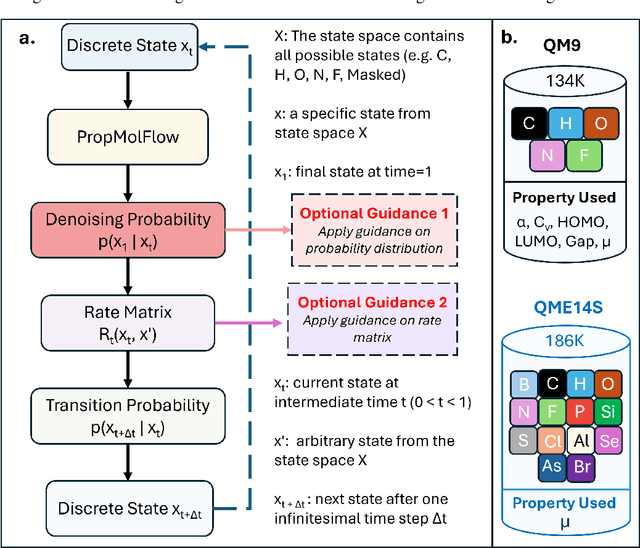
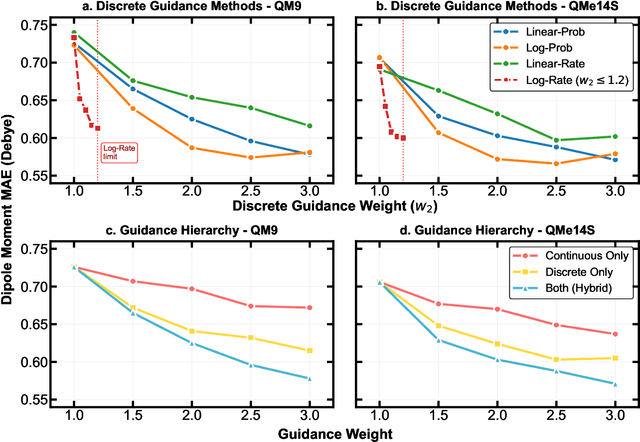
Key objectives in conditional molecular generation include ensuring chemical validity, aligning generated molecules with target properties, promoting structural diversity, and enabling efficient sampling for discovery. Recent advances in computer vision introduced a range of new guidance strategies for generative models, many of which can be adapted to support these goals. In this work, we integrate state-of-the-art guidance methods -- including classifier-free guidance, autoguidance, and model guidance -- in a leading molecule generation framework built on an SE(3)-equivariant flow matching process. We propose a hybrid guidance strategy that separately guides continuous and discrete molecular modalities -- operating on velocity fields and predicted logits, respectively -- while jointly optimizing their guidance scales via Bayesian optimization. Our implementation, benchmarked on the QM9 and QMe14S datasets, achieves new state-of-the-art performance in property alignment for de novo molecular generation. The generated molecules also exhibit high structural validity. Furthermore, we systematically compare the strengths and limitations of various guidance methods, offering insights into their broader applicability.
Contrastive Self-Supervised Learning As Neural Manifold Packing
Jun 16, 2025Contrastive self-supervised learning based on point-wise comparisons has been widely studied for vision tasks. In the visual cortex of the brain, neuronal responses to distinct stimulus classes are organized into geometric structures known as neural manifolds. Accurate classification of stimuli can be achieved by effectively separating these manifolds, akin to solving a packing problem. We introduce Contrastive Learning As Manifold Packing (CLAMP), a self-supervised framework that recasts representation learning as a manifold packing problem. CLAMP introduces a loss function inspired by the potential energy of short-range repulsive particle systems, such as those encountered in the physics of simple liquids and jammed packings. In this framework, each class consists of sub-manifolds embedding multiple augmented views of a single image. The sizes and positions of the sub-manifolds are dynamically optimized by following the gradient of a packing loss. This approach yields interpretable dynamics in the embedding space that parallel jamming physics, and introduces geometrically meaningful hyperparameters within the loss function. Under the standard linear evaluation protocol, which freezes the backbone and trains only a linear classifier, CLAMP achieves competitive performance with state-of-the-art self-supervised models. Furthermore, our analysis reveals that neural manifolds corresponding to different categories emerge naturally and are effectively separated in the learned representation space, highlighting the potential of CLAMP to bridge insights from physics, neural science, and machine learning.
A practical guide to machine learning interatomic potentials -- Status and future
Mar 12, 2025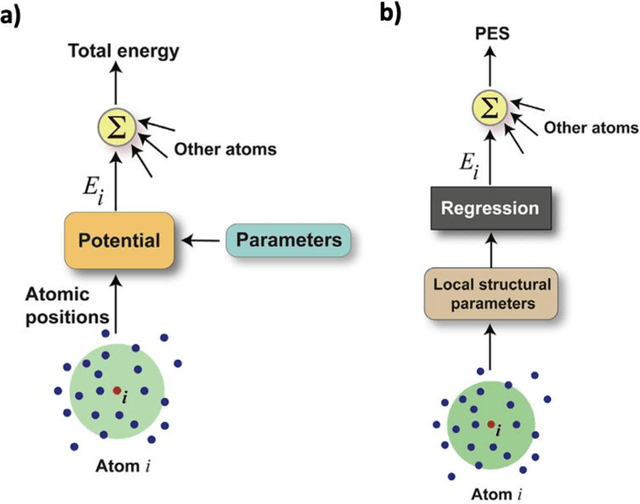

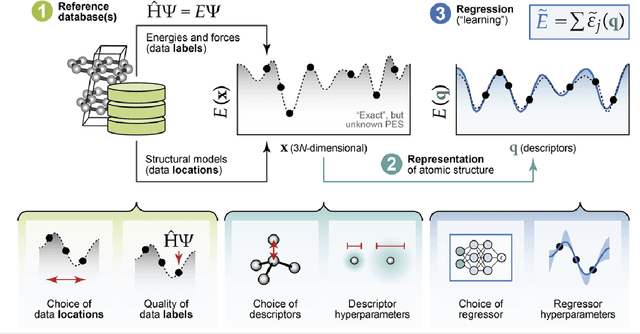
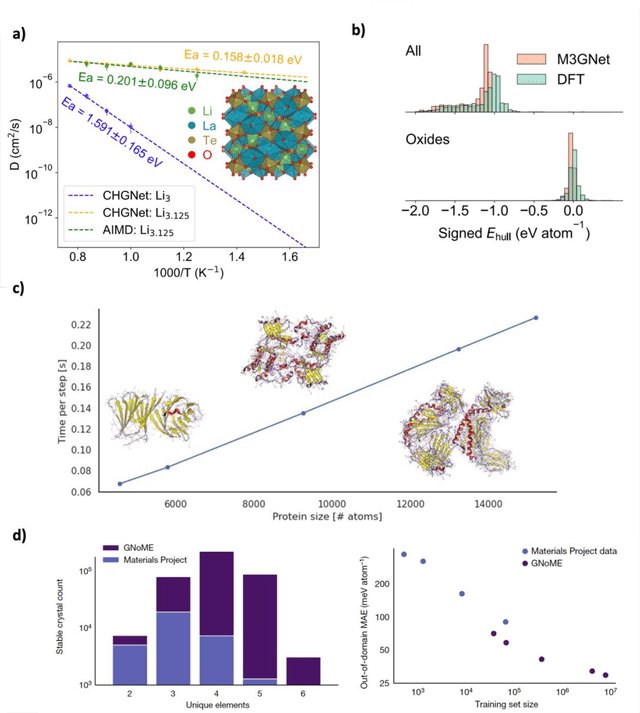
The rapid development and large body of literature on machine learning interatomic potentials (MLIPs) can make it difficult to know how to proceed for researchers who are not experts but wish to use these tools. The spirit of this review is to help such researchers by serving as a practical, accessible guide to the state-of-the-art in MLIPs. This review paper covers a broad range of topics related to MLIPs, including (i) central aspects of how and why MLIPs are enablers of many exciting advancements in molecular modeling, (ii) the main underpinnings of different types of MLIPs, including their basic structure and formalism, (iii) the potentially transformative impact of universal MLIPs for both organic and inorganic systems, including an overview of the most recent advances, capabilities, downsides, and potential applications of this nascent class of MLIPs, (iv) a practical guide for estimating and understanding the execution speed of MLIPs, including guidance for users based on hardware availability, type of MLIP used, and prospective simulation size and time, (v) a manual for what MLIP a user should choose for a given application by considering hardware resources, speed requirements, energy and force accuracy requirements, as well as guidance for choosing pre-trained potentials or fitting a new potential from scratch, (vi) discussion around MLIP infrastructure, including sources of training data, pre-trained potentials, and hardware resources for training, (vii) summary of some key limitations of present MLIPs and current approaches to mitigate such limitations, including methods of including long-range interactions, handling magnetic systems, and treatment of excited states, and finally (viii) we finish with some more speculative thoughts on what the future holds for the development and application of MLIPs over the next 3-10+ years.
Open Materials Generation with Stochastic Interpolants
Feb 04, 2025The discovery of new materials is essential for enabling technological advancements. Computational approaches for predicting novel materials must effectively learn the manifold of stable crystal structures within an infinite design space. We introduce Open Materials Generation (OMG), a unifying framework for the generative design and discovery of inorganic crystalline materials. OMG employs stochastic interpolants (SI) to bridge an arbitrary base distribution to the target distribution of inorganic crystals via a broad class of tunable stochastic processes, encompassing both diffusion models and flow matching as special cases. In this work, we adapt the SI framework by integrating an equivariant graph representation of crystal structures and extending it to account for periodic boundary conditions in unit cell representations. Additionally, we couple the SI flow over spatial coordinates and lattice vectors with discrete flow matching for atomic species. We benchmark OMG's performance on two tasks: Crystal Structure Prediction (CSP) for specified compositions, and 'de novo' generation (DNG) aimed at discovering stable, novel, and unique structures. In our ground-up implementation of OMG, we refine and extend both CSP and DNG metrics compared to previous works. OMG establishes a new state-of-the-art in generative modeling for materials discovery, outperforming purely flow-based and diffusion-based implementations. These results underscore the importance of designing flexible deep learning frameworks to accelerate progress in materials science.
Unconditional stability of a recurrent neural circuit implementing divisive normalization
Sep 27, 2024Stability in recurrent neural models poses a significant challenge, particularly in developing biologically plausible neurodynamical models that can be seamlessly trained. Traditional cortical circuit models are notoriously difficult to train due to expansive nonlinearities in the dynamical system, leading to an optimization problem with nonlinear stability constraints that are difficult to impose. Conversely, recurrent neural networks (RNNs) excel in tasks involving sequential data but lack biological plausibility and interpretability. In this work, we address these challenges by linking dynamic divisive normalization (DN) to the stability of ORGaNICs, a biologically plausible recurrent cortical circuit model that dynamically achieves DN and has been shown to simulate a wide range of neurophysiological phenomena. By using the indirect method of Lyapunov, we prove the remarkable property of unconditional local stability for an arbitrary-dimensional ORGaNICs circuit when the recurrent weight matrix is the identity. We thus connect ORGaNICs to a system of coupled damped harmonic oscillators, which enables us to derive the circuit's energy function, providing a normative principle of what the circuit, and individual neurons, aim to accomplish. Further, for a generic recurrent weight matrix, we prove the stability of the 2D model and demonstrate empirically that stability holds in higher dimensions. Finally, we show that ORGaNICs can be trained by backpropagation through time without gradient clipping/scaling, thanks to its intrinsic stability property and adaptive time constants, which address the problems of exploding, vanishing, and oscillating gradients. By evaluating the model's performance on RNN benchmarks, we find that ORGaNICs outperform alternative neurodynamical models on static image classification tasks and perform comparably to LSTMs on sequential tasks.
On the design space between molecular mechanics and machine learning force fields
Sep 03, 2024A force field as accurate as quantum mechanics (QM) and as fast as molecular mechanics (MM), with which one can simulate a biomolecular system efficiently enough and meaningfully enough to get quantitative insights, is among the most ardent dreams of biophysicists -- a dream, nevertheless, not to be fulfilled any time soon. Machine learning force fields (MLFFs) represent a meaningful endeavor towards this direction, where differentiable neural functions are parametrized to fit ab initio energies, and furthermore forces through automatic differentiation. We argue that, as of now, the utility of the MLFF models is no longer bottlenecked by accuracy but primarily by their speed (as well as stability and generalizability), as many recent variants, on limited chemical spaces, have long surpassed the chemical accuracy of $1$ kcal/mol -- the empirical threshold beyond which realistic chemical predictions are possible -- though still magnitudes slower than MM. Hoping to kindle explorations and designs of faster, albeit perhaps slightly less accurate MLFFs, in this review, we focus our attention on the design space (the speed-accuracy tradeoff) between MM and ML force fields. After a brief review of the building blocks of force fields of either kind, we discuss the desired properties and challenges now faced by the force field development community, survey the efforts to make MM force fields more accurate and ML force fields faster, envision what the next generation of MLFF might look like.