 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOn the design space between molecular mechanics and machine learning force fields
Sep 03, 2024A force field as accurate as quantum mechanics (QM) and as fast as molecular mechanics (MM), with which one can simulate a biomolecular system efficiently enough and meaningfully enough to get quantitative insights, is among the most ardent dreams of biophysicists -- a dream, nevertheless, not to be fulfilled any time soon. Machine learning force fields (MLFFs) represent a meaningful endeavor towards this direction, where differentiable neural functions are parametrized to fit ab initio energies, and furthermore forces through automatic differentiation. We argue that, as of now, the utility of the MLFF models is no longer bottlenecked by accuracy but primarily by their speed (as well as stability and generalizability), as many recent variants, on limited chemical spaces, have long surpassed the chemical accuracy of $1$ kcal/mol -- the empirical threshold beyond which realistic chemical predictions are possible -- though still magnitudes slower than MM. Hoping to kindle explorations and designs of faster, albeit perhaps slightly less accurate MLFFs, in this review, we focus our attention on the design space (the speed-accuracy tradeoff) between MM and ML force fields. After a brief review of the building blocks of force fields of either kind, we discuss the desired properties and challenges now faced by the force field development community, survey the efforts to make MM force fields more accurate and ML force fields faster, envision what the next generation of MLFF might look like.
An Extendible, Graph-Neural-Network-Based Approach for Accurate Force Field Development of Large Flexible Organic Molecules
Jun 02, 2021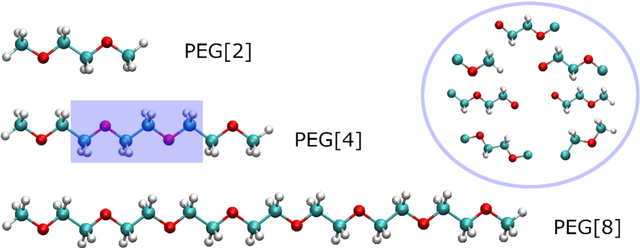

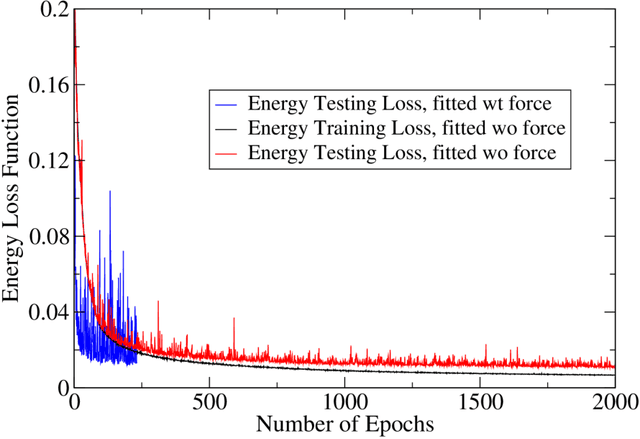
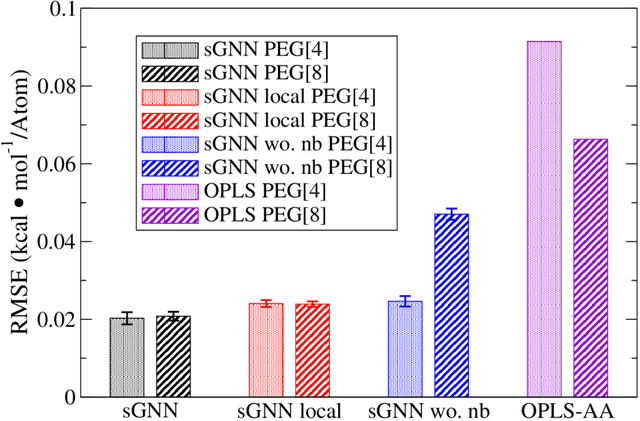
An accurate force field is the key to the success of all molecular mechanics simulations on organic polymers and biomolecules. Accuracy beyond density functional theory is often needed to describe the intermolecular interactions, while most correlated wavefunction (CW) methods are prohibitively expensive for large molecules. Therefore, it posts a great challenge to develop an extendible ab initio force field for large flexible organic molecules at CW level of accuracy. In this work, we face this challenge by combining the physics-driven nonbonding potential with a data-driven subgraph neural network bonding model (named sGNN). Tests on polyethylene glycol polymer chains show that our strategy is highly accurate and robust for molecules of different sizes. Therefore, we can develop the force field from small molecular fragments (with sizes easily accessible to CW methods) and safely transfer it to large polymers, thus opening a new path to the next-generation organic force fields.