 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMAD: Microenvironment-Aware Distillation -- A Pretraining Strategy for Virtual Spatial Omics from Microscopy
Mar 11, 2026Bridging microscopy and omics would allow us to read molecular states from images-at single-cell resolution and tissue scale-without the cost and throughput limits of omics technologies. Self-supervised pretraining offers a scalable approach with minimal labels, yet how to encode single-cell identity within tissue environments-and the extent of biological information such models can capture-remains an open question. Here, we introduce MAD (microenvironment-aware distillation), a pretraining strategy that learns cell-centric embeddings by jointly self-distilling the morphology view and the microenvironment view of the same indexed cell into a unified embedding space. Across diverse tissues and imaging modalities, MAD achieves state-of-the-art prediction performance on downstream tasks including cell subtyping, transcriptomic prediction, and bioinformatic inference. MAD even outperforms foundation models with a similar number of model parameters that have been trained on substantially larger datasets. These results demonstrate that MAD's dual-view joint self-distillation effectively captures the complexity and diversity of cells within tissues. Together, this establishes MAD as a general tool for representation learning in microscopy, enabling virtual spatial omics and biological insights from vast microscopy datasets.
Otter: Generating Tests from Issues to Validate SWE Patches
Feb 07, 2025While there has been plenty of work on generating tests from existing code, there has been limited work on generating tests from issues. A correct test must validate the code patch that resolves the issue. In this work, we focus on the scenario where the code patch does not exist yet. This approach supports two major use-cases. First, it supports TDD (test-driven development), the discipline of "test first, write code later" that has well-documented benefits for human software engineers. Second, it also validates SWE (software engineering) agents, which generate code patches for resolving issues. This paper introduces Otter, an LLM-based solution for generating tests from issues. Otter augments LLMs with rule-based analysis to check and repair their outputs, and introduces a novel self-reflective action planning stage. Experiments show Otter outperforming state-of-the-art systems for generating tests from issues, in addition to enhancing systems that generate patches from issues. We hope that Otter helps make developers more productive at resolving issues and leads to more robust, well-tested code.
AutoRestTest: A Tool for Automated REST API Testing Using LLMs and MARL
Jan 15, 2025As REST APIs have become widespread in modern web services, comprehensive testing of these APIs has become increasingly crucial. Due to the vast search space consisting of operations, parameters, and parameter values along with their complex dependencies and constraints, current testing tools suffer from low code coverage, leading to suboptimal fault detection. To address this limitation, we present a novel tool, AutoRestTest, which integrates the Semantic Operation Dependency Graph (SODG) with Multi-Agent Reinforcement Learning (MARL) and large language models (LLMs) for effective REST API testing. AutoRestTest determines operation-dependent parameters using the SODG and employs five specialized agents (operation, parameter, value, dependency, and header) to identify dependencies of operations and generate operation sequences, parameter combinations, and values. AutoRestTest provides a command-line interface and continuous telemetry on successful operation count, unique server errors detected, and time elapsed. Upon completion, AutoRestTest generates a detailed report highlighting errors detected and operations exercised. In this paper, we introduce our tool and present preliminary results.
LlamaRestTest: Effective REST API Testing with Small Language Models
Jan 15, 2025
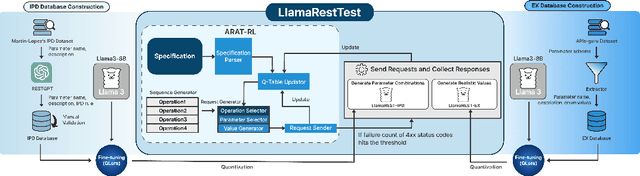
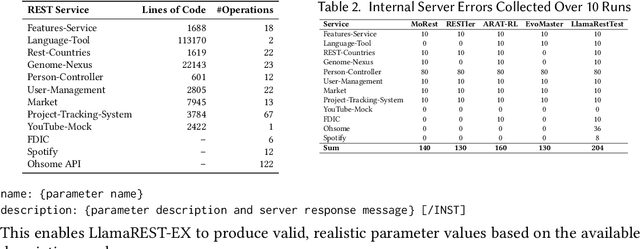
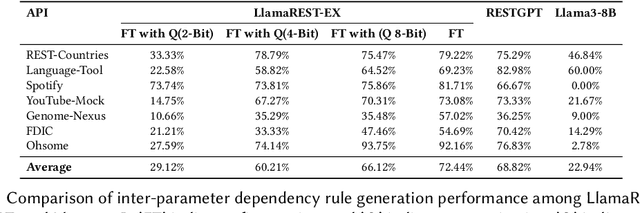
Modern web services rely heavily on REST APIs, typically documented using the OpenAPI specification. The widespread adoption of this standard has resulted in the development of many black-box testing tools that generate tests based on these specifications. Recent advancements in Natural Language Processing (NLP), particularly with Large Language Models (LLMs), have enhanced REST API testing by extracting actionable rules and generating input values from the human-readable portions of the specification. However, these advancements overlook the potential of continuously refining the identified rules and test inputs based on server responses. To address this limitation, we present LlamaRestTest, a novel approach that employs two custom LLMs to generate realistic test inputs and uncover parameter dependencies during the testing process by incorporating server responses. These LLMs are created by fine-tuning the Llama3-8b model, using mined datasets of REST API example values and inter-parameter dependencies. We evaluated LlamaRestTest on 12 real-world services (including popular services such as Spotify), comparing it against RESTGPT, a GPT-powered specification-enhancement tool, as well as several state-of-the-art REST API testing tools, including RESTler, MoRest, EvoMaster, and ARAT-RL. Our results show that fine-tuning enables smaller LLMs to outperform larger models in detecting actionable rules and generating inputs for REST API testing. We evaluated configurations from the base Llama3-8B to fine-tuned versions and explored 2-bit, 4-bit, and 8-bit quantization for efficiency. LlamaRestTest surpasses state-of-the-art tools in code coverage and error detection, even with RESTGPT-enhanced specifications, and an ablation study highlights the impact of its novel components.
TDD-Bench Verified: Can LLMs Generate Tests for Issues Before They Get Resolved?
Dec 03, 2024



Test-driven development (TDD) is the practice of writing tests first and coding later, and the proponents of TDD expound its numerous benefits. For instance, given an issue on a source code repository, tests can clarify the desired behavior among stake-holders before anyone writes code for the agreed-upon fix. Although there has been a lot of work on automated test generation for the practice "write code first, test later", there has been little such automation for TDD. Ideally, tests for TDD should be fail-to-pass (i.e., fail before the issue is resolved and pass after) and have good adequacy with respect to covering the code changed during issue resolution. This paper introduces TDD-Bench Verified, a high-quality benchmark suite of 449 issues mined from real-world GitHub code repositories. The benchmark's evaluation harness runs only relevant tests in isolation for simple yet accurate coverage measurements, and the benchmark's dataset is filtered both by human judges and by execution in the harness. This paper also presents Auto-TDD, an LLM-based solution that takes as input an issue description and a codebase (prior to issue resolution) and returns as output a test that can be used to validate the changes made for resolving the issue. Our evaluation shows that Auto-TDD yields a better fail-to-pass rate than the strongest prior work while also yielding high coverage adequacy. Overall, we hope that this work helps make developers more productive at resolving issues while simultaneously leading to more robust fixes.
A Multi-Agent Approach for REST API Testing with Semantic Graphs and LLM-Driven Inputs
Nov 11, 2024As modern web services increasingly rely on REST APIs, their thorough testing has become crucial. Furthermore, the advent of REST API specifications such as the OpenAPI Specification has led to the emergence of many black-box REST API testing tools. However, these tools often focus on individual test elements in isolation (e.g., APIs, parameters, values), resulting in lower coverage and less effectiveness in detecting faults (i.e., 500 response codes). To address these limitations, we present AutoRestTest, the first black-box framework to adopt a dependency-embedded multi-agent approach for REST API testing, integrating Multi-Agent Reinforcement Learning (MARL) with a Semantic Property Dependency Graph (SPDG) and Large Language Models (LLMs). Our approach treats REST API testing as a separable problem, where four agents -- API, dependency, parameter, and value -- collaborate to optimize API exploration. LLMs handle domain-specific value restrictions, the SPDG model simplifies the search space for dependencies using a similarity score between API operations, and MARL dynamically optimizes the agents' behavior. Evaluated on 12 real-world REST services, AutoRestTest outperforms the four leading black-box REST API testing tools, including those assisted by RESTGPT (which augments realistic test inputs using LLMs), in terms of code coverage, operation coverage, and fault detection. Notably, AutoRestTest is the only tool able to identify an internal server error in Spotify. Our ablation study underscores the significant contributions of the agent learning, SPDG, and LLM components.
Repository-Level Compositional Code Translation and Validation
Oct 31, 2024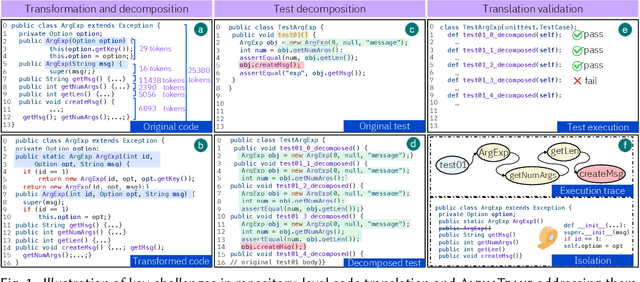
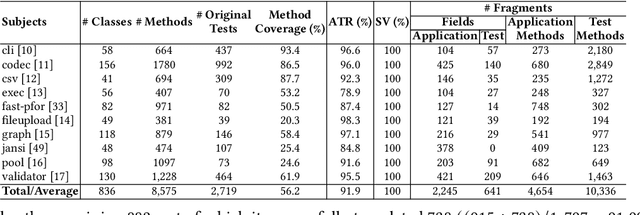
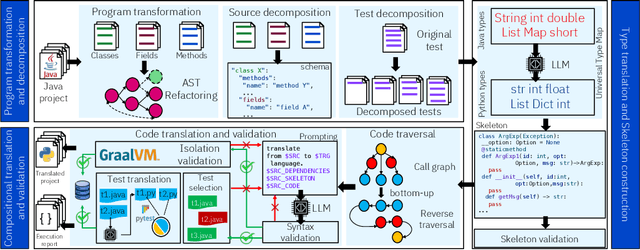
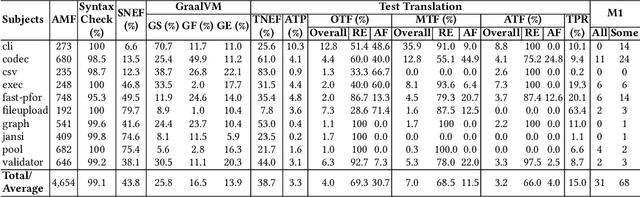
Code translation transforms programs from one programming language (PL) to another. Several rule-based transpilers have been designed to automate code translation between different pairs of PLs. However, the rules can become obsolete as the PLs evolve and cannot generalize to other PLs. Recent studies have explored the automation of code translation using Large Language Models (LLMs). One key observation is that such techniques may work well for crafted benchmarks but fail to generalize to the scale and complexity of real-world projects with dependencies, custom types, PL-specific features, etc. We propose AlphaTrans, a neuro-symbolic approach to automate repository-level code translation. AlphaTrans translates both source and test code, and employs multiple levels of validation to ensure the translation preserves the functionality of the source program. To break down the problem for LLMs, AlphaTrans leverages program analysis to decompose the program into fragments and translates them in the reverse call order. We leveraged AlphaTrans to translate ten real-world open-source projects consisting of <836, 8575, 2719> classes, methods, and tests. AlphaTrans translated the entire repository of these projects consisting of 6899 source code fragments. 99.1% of the translated code fragments are syntactically correct, and AlphaTrans validates the translations' runtime behavior and functional correctness for 25.8%. On average, the integrated translation and validation take 36 hours to translate a project, showing its scalability in practice. For the syntactically or semantically incorrect translations, AlphaTrans generates a report including existing translation, stack trace, test errors, or assertion failures. We provided these artifacts to two developers to fix the translation bugs in four projects. They were able to fix the issues in 20.1 hours on average and achieve all passing tests.
Property-Guided Generative Modelling for Robust Model-Based Design with Imbalanced Data
May 23, 2023



The problem of designing protein sequences with desired properties is challenging, as it requires to explore a high-dimensional protein sequence space with extremely sparse meaningful regions. This has led to the development of model-based optimization (MBO) techniques that aid in the design, by using effective search models guided by the properties over the sequence space. However, the intrinsic imbalanced nature of experimentally derived datasets causes existing MBO approaches to struggle or outright fail. We propose a property-guided variational auto-encoder (PGVAE) whose latent space is explicitly structured by the property values such that samples are prioritized according to these properties. Through extensive benchmarking on real and semi-synthetic protein datasets, we demonstrate that MBO with PGVAE robustly finds sequences with improved properties despite significant dataset imbalances. We further showcase the generality of our approach to continuous design spaces, and its robustness to dataset imbalance in an application to physics-informed neural networks.
CIMLA: Interpretable AI for inference of differential causal networks
Apr 25, 2023The discovery of causal relationships from high-dimensional data is a major open problem in bioinformatics. Machine learning and feature attribution models have shown great promise in this context but lack causal interpretation. Here, we show that a popular feature attribution model estimates a causal quantity reflecting the influence of one variable on another, under certain assumptions. We leverage this insight to implement a new tool, CIMLA, for discovering condition-dependent changes in causal relationships. We then use CIMLA to identify differences in gene regulatory networks between biological conditions, a problem that has received great attention in recent years. Using extensive benchmarking on simulated data sets, we show that CIMLA is more robust to confounding variables and is more accurate than leading methods. Finally, we employ CIMLA to analyze a previously published single-cell RNA-seq data set collected from subjects with and without Alzheimer's disease (AD), discovering several potential regulators of AD.
Toward Scalable Machine Learning and Data Mining: the Bioinformatics Case
Sep 29, 2017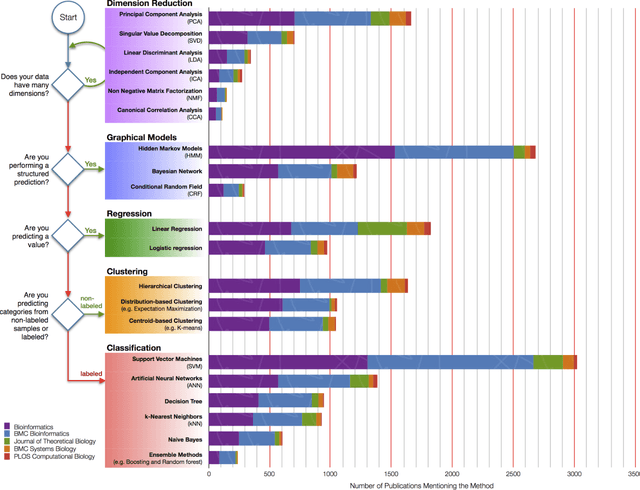
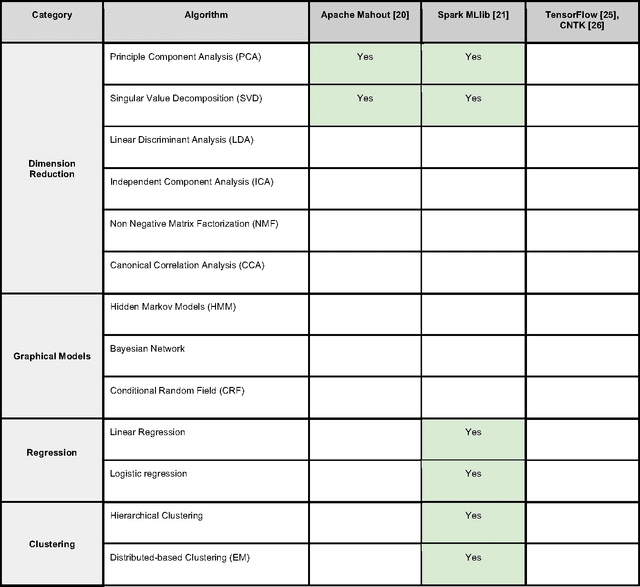
In an effort to overcome the data deluge in computational biology and bioinformatics and to facilitate bioinformatics research in the era of big data, we identify some of the most influential algorithms that have been widely used in the bioinformatics community. These top data mining and machine learning algorithms cover classification, clustering, regression, graphical model-based learning, and dimensionality reduction. The goal of this study is to guide the focus of scalable computing experts in the endeavor of applying new storage and scalable computation designs to bioinformatics algorithms that merit their attention most, following the engineering maxim of "optimize the common case".