 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgemCLM: A Function-Infused and Synthesis-Friendly Modular Chemical Language Model
May 18, 2025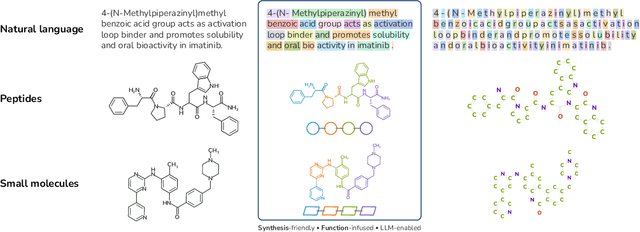
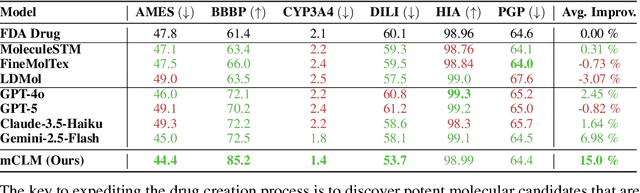
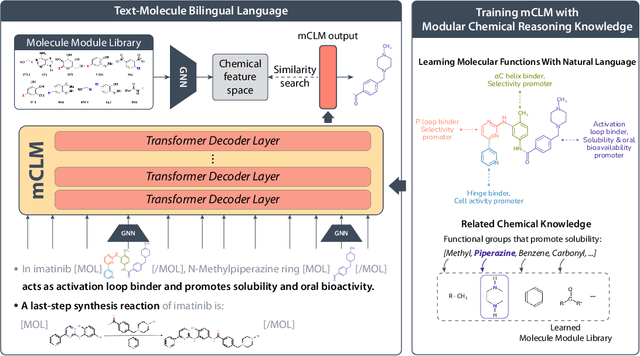
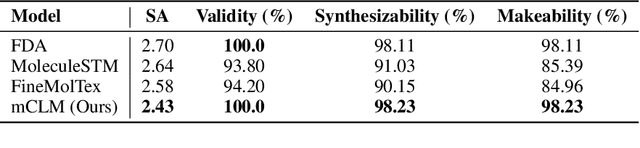
Despite their ability to understand chemical knowledge and accurately generate sequential representations, large language models (LLMs) remain limited in their capacity to propose novel molecules with drug-like properties. In addition, the molecules that LLMs propose can often be challenging to make in the lab. To more effectively enable the discovery of functional small molecules, LLMs need to learn a molecular language. However, LLMs are currently limited by encoding molecules from atoms. In this paper, we argue that just like tokenizing texts into (sub-)word tokens instead of characters, molecules should be decomposed and reassembled at the level of functional building blocks, i.e., parts of molecules that bring unique functions and serve as effective building blocks for real-world automated laboratory synthesis. This motivates us to propose mCLM, a modular Chemical-Language Model tokenizing molecules into building blocks and learning a bilingual language model of both natural language descriptions of functions and molecule building blocks. By reasoning on such functional building blocks, mCLM guarantees to generate efficiently synthesizable molecules thanks to recent progress in block-based chemistry, while also improving the functions of molecules in a principled manner. In experiments on 430 FDA-approved drugs, we find mCLM capable of significantly improving 5 out of 6 chemical functions critical to determining drug potentials. More importantly, mCLM can reason on multiple functions and improve the FDA-rejected drugs (``fallen angels'') over multiple iterations to greatly improve their shortcomings.
FARM: Functional Group-Aware Representations for Small Molecules
Oct 02, 2024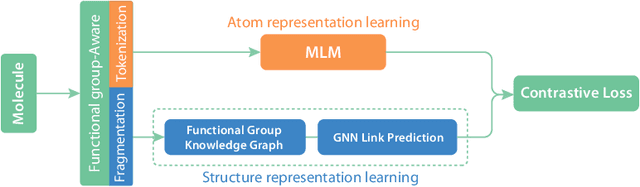

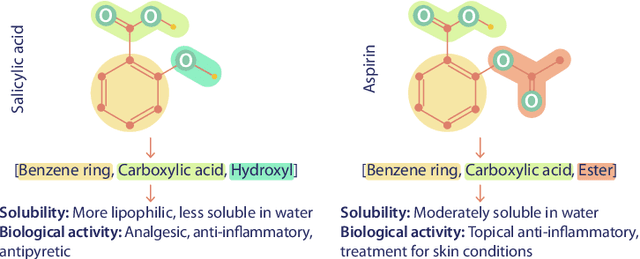
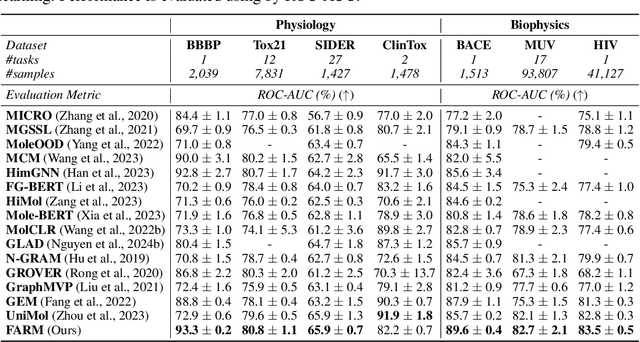
We introduce Functional Group-Aware Representations for Small Molecules (FARM), a novel foundation model designed to bridge the gap between SMILES, natural language, and molecular graphs. The key innovation of FARM lies in its functional group-aware tokenization, which incorporates functional group information directly into the representations. This strategic reduction in tokenization granularity in a way that is intentionally interfaced with key drivers of functional properties (i.e., functional groups) enhances the model's understanding of chemical language, expands the chemical lexicon, more effectively bridging SMILES and natural language, and ultimately advances the model's capacity to predict molecular properties. FARM also represents molecules from two perspectives: by using masked language modeling to capture atom-level features and by employing graph neural networks to encode the whole molecule topology. By leveraging contrastive learning, FARM aligns these two views of representations into a unified molecular embedding. We rigorously evaluate FARM on the MoleculeNet dataset, where it achieves state-of-the-art performance on 10 out of 12 tasks. These results highlight FARM's potential to improve molecular representation learning, with promising applications in drug discovery and pharmaceutical research.
Property-Guided Generative Modelling for Robust Model-Based Design with Imbalanced Data
May 23, 2023



The problem of designing protein sequences with desired properties is challenging, as it requires to explore a high-dimensional protein sequence space with extremely sparse meaningful regions. This has led to the development of model-based optimization (MBO) techniques that aid in the design, by using effective search models guided by the properties over the sequence space. However, the intrinsic imbalanced nature of experimentally derived datasets causes existing MBO approaches to struggle or outright fail. We propose a property-guided variational auto-encoder (PGVAE) whose latent space is explicitly structured by the property values such that samples are prioritized according to these properties. Through extensive benchmarking on real and semi-synthetic protein datasets, we demonstrate that MBO with PGVAE robustly finds sequences with improved properties despite significant dataset imbalances. We further showcase the generality of our approach to continuous design spaces, and its robustness to dataset imbalance in an application to physics-informed neural networks.
Chemical-Reaction-Aware Molecule Representation Learning
Sep 22, 2021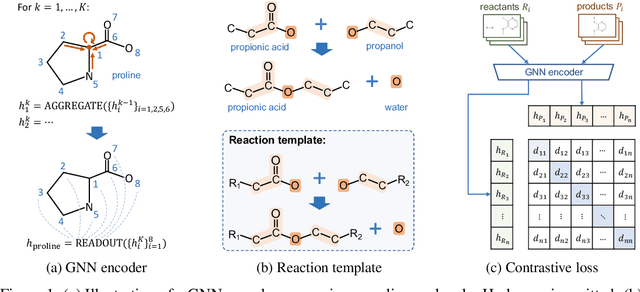
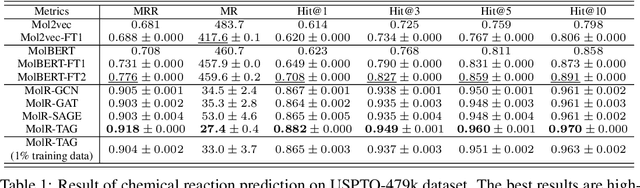
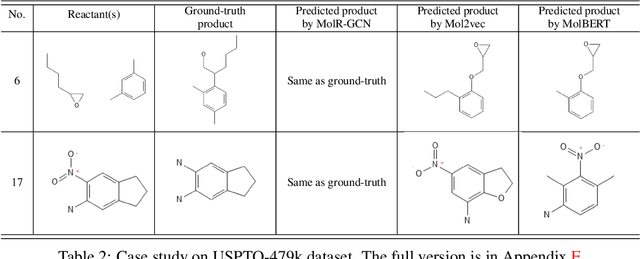
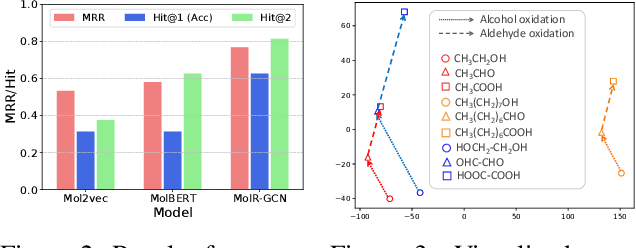
Molecule representation learning (MRL) methods aim to embed molecules into a real vector space. However, existing SMILES-based (Simplified Molecular-Input Line-Entry System) or GNN-based (Graph Neural Networks) MRL methods either take SMILES strings as input that have difficulty in encoding molecule structure information, or over-emphasize the importance of GNN architectures but neglect their generalization ability. Here we propose using chemical reactions to assist learning molecule representation. The key idea of our approach is to preserve the equivalence of molecules with respect to chemical reactions in the embedding space, i.e., forcing the sum of reactant embeddings and the sum of product embeddings to be equal for each chemical equation. This constraint is proven effective to 1) keep the embedding space well-organized and 2) improve the generalization ability of molecule embeddings. Moreover, our model can use any GNN as the molecule encoder and is thus agnostic to GNN architectures. Experimental results demonstrate that our method achieves state-of-the-art performance in a variety of downstream tasks, e.g., 17.4% absolute Hit@1 gain in chemical reaction prediction, 2.3% absolute AUC gain in molecule property prediction, and 18.5% relative RMSE gain in graph-edit-distance prediction, respectively, over the best baseline method. The code is available at https://github.com/hwwang55/MolR.