 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDo LLMs Build Spatial World Models? Evidence from Grid-World Maze Tasks
Apr 12, 2026Foundation models have shown remarkable performance across diverse tasks, yet their ability to construct internal spatial world models for reasoning and planning remains unclear. We systematically evaluate the spatial understanding of large language models through maze tasks, a controlled testing context requiring multi-step planning and spatial abstraction. Across comprehensive experiments with Gemini-2.5-Flash, GPT-5-mini, Claude-Haiku-4.5, and DeepSeek-Chat, we uncover significant discrepancies in spatial reasoning that challenge assumptions about LLM planning capabilities. Using chain-of-thought prompting, Gemini achieves 80-86% accuracy on smaller mazes (5x5 to 7x7 grids) with tokenized adjacency representations, but performance collapses to 16-34% with visual grid formats, which is a 2-5x difference, suggesting representation-dependent rather than format-invariant spatial reasoning. We further probe spatial understanding through sequential proximity questions and compositional distance comparisons. Despite achieving 96-99% semantic coverage in reasoning traces, models fail to leverage this understanding for consistent spatial computations, indicating that they treat each question independently rather than building cumulative spatial knowledge. Our findings based on the maze-solving tasks suggest that LLMs do not develop robust spatial world models, but rather exhibit representation-specific and prompting-dependent reasoning that succeeds only under narrow conditions. These results have critical implications for deploying foundation models in applications requiring spatial abstraction.
Emotion-cause pair extraction method based on multi-granularity information and multi-module interaction
Apr 10, 2024The purpose of emotion-cause pair extraction is to extract the pair of emotion clauses and cause clauses. On the one hand, the existing methods do not take fully into account the relationship between the emotion extraction of two auxiliary tasks. On the other hand, the existing two-stage model has the problem of error propagation. In addition, existing models do not adequately address the emotion and cause-induced locational imbalance of samples. To solve these problems, an end-to-end multitasking model (MM-ECPE) based on shared interaction between GRU, knowledge graph and transformer modules is proposed. Furthermore, based on MM-ECPE, in order to use the encoder layer to better solve the problem of imbalanced distribution of clause distances between clauses and emotion clauses, we propose a novel encoding based on BERT, sentiment lexicon, and position-aware interaction module layer of emotion motif pair retrieval model (MM-ECPE(BERT)). The model first fully models the interaction between different tasks through the multi-level sharing module, and mines the shared information between emotion-cause pair extraction and the emotion extraction and cause extraction. Second, to solve the imbalanced distribution of emotion clauses and cause clauses problem, suitable labels are screened out according to the knowledge graph path length and task-specific features are constructed so that the model can focus on extracting pairs with corresponding emotion-cause relationships. Experimental results on the ECPE benchmark dataset show that the proposed model achieves good performance, especially on position-imbalanced samples.
Towards out-of-distribution generalizable predictions of chemical kinetics properties
Oct 04, 2023



Machine Learning (ML) techniques have found applications in estimating chemical kinetics properties. With the accumulated drug molecules identified through "AI4drug discovery", the next imperative lies in AI-driven design for high-throughput chemical synthesis processes, with the estimation of properties of unseen reactions with unexplored molecules. To this end, the existing ML approaches for kinetics property prediction are required to be Out-Of-Distribution (OOD) generalizable. In this paper, we categorize the OOD kinetic property prediction into three levels (structure, condition, and mechanism), revealing unique aspects of such problems. Under this framework, we create comprehensive datasets to benchmark (1) the state-of-the-art ML approaches for reaction prediction in the OOD setting and (2) the state-of-the-art graph OOD methods in kinetics property prediction problems. Our results demonstrated the challenges and opportunities in OOD kinetics property prediction. Our datasets and benchmarks can further support research in this direction.
Chemical-Reaction-Aware Molecule Representation Learning
Sep 22, 2021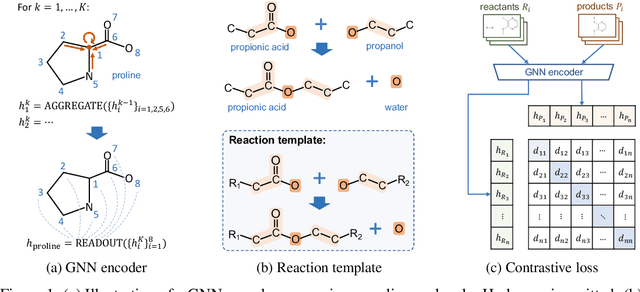
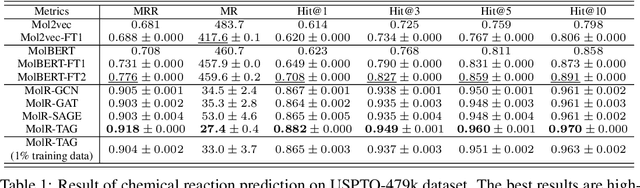
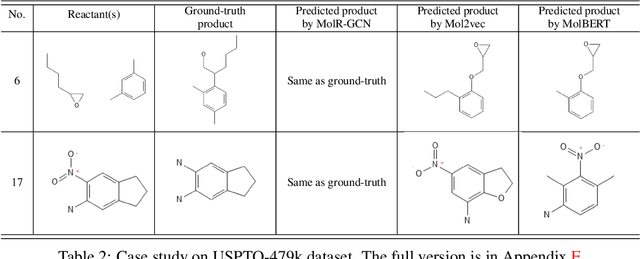
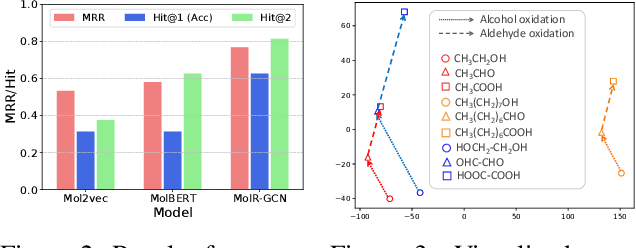
Molecule representation learning (MRL) methods aim to embed molecules into a real vector space. However, existing SMILES-based (Simplified Molecular-Input Line-Entry System) or GNN-based (Graph Neural Networks) MRL methods either take SMILES strings as input that have difficulty in encoding molecule structure information, or over-emphasize the importance of GNN architectures but neglect their generalization ability. Here we propose using chemical reactions to assist learning molecule representation. The key idea of our approach is to preserve the equivalence of molecules with respect to chemical reactions in the embedding space, i.e., forcing the sum of reactant embeddings and the sum of product embeddings to be equal for each chemical equation. This constraint is proven effective to 1) keep the embedding space well-organized and 2) improve the generalization ability of molecule embeddings. Moreover, our model can use any GNN as the molecule encoder and is thus agnostic to GNN architectures. Experimental results demonstrate that our method achieves state-of-the-art performance in a variety of downstream tasks, e.g., 17.4% absolute Hit@1 gain in chemical reaction prediction, 2.3% absolute AUC gain in molecule property prediction, and 18.5% relative RMSE gain in graph-edit-distance prediction, respectively, over the best baseline method. The code is available at https://github.com/hwwang55/MolR.
Fine-Grained Chemical Entity Typing with Multimodal Knowledge Representation
Aug 29, 2021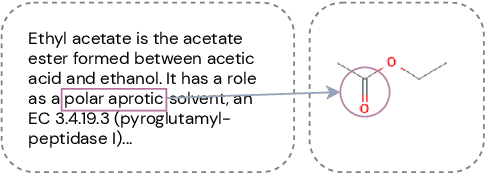
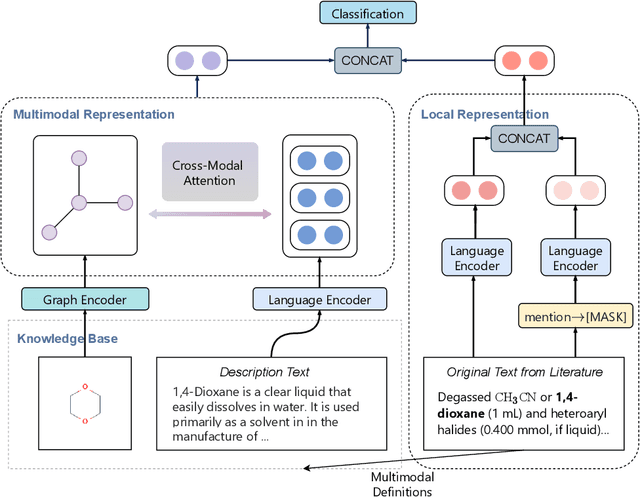
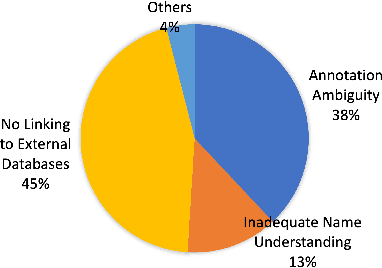
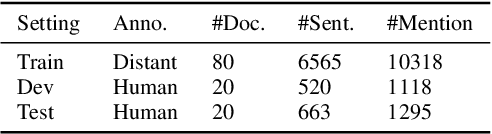
Automated knowledge discovery from trending chemical literature is essential for more efficient biomedical research. How to extract detailed knowledge about chemical reactions from the core chemistry literature is a new emerging challenge that has not been well studied. In this paper, we study the new problem of fine-grained chemical entity typing, which poses interesting new challenges especially because of the complex name mentions frequently occurring in chemistry literature and graphic representation of entities. We introduce a new benchmark data set (CHEMET) to facilitate the study of the new task and propose a novel multi-modal representation learning framework to solve the problem of fine-grained chemical entity typing by leveraging external resources with chemical structures and using cross-modal attention to learn effective representation of text in the chemistry domain. Experiment results show that the proposed framework outperforms multiple state-of-the-art methods.