 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Topic Modeling Analysis of Stigma Dimensions, Social, and Related Behavioral Circumstances in Clinical Notes Among Patients with HIV
Jun 10, 2025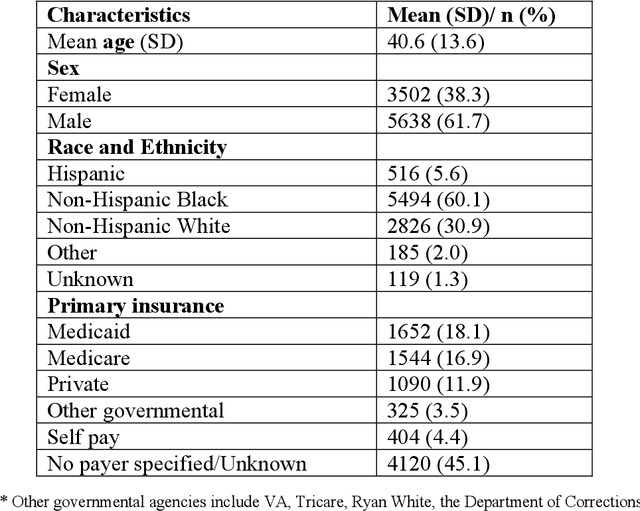
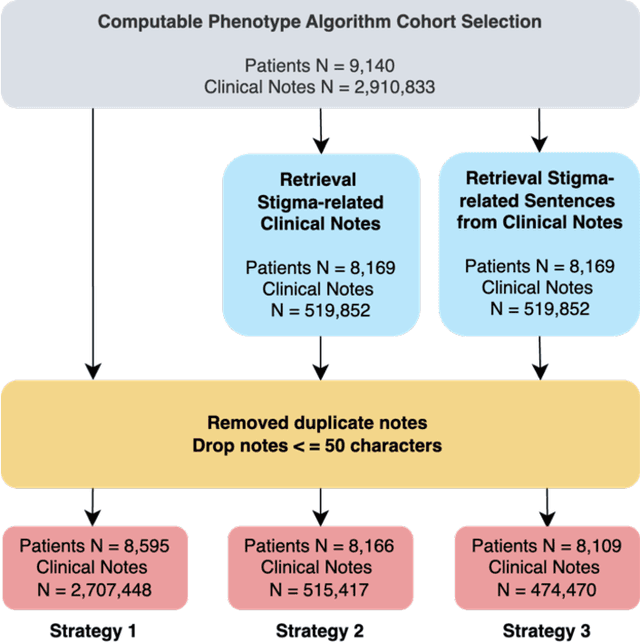
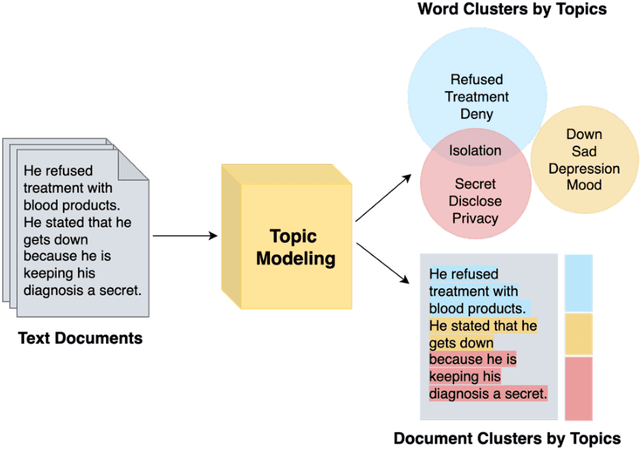
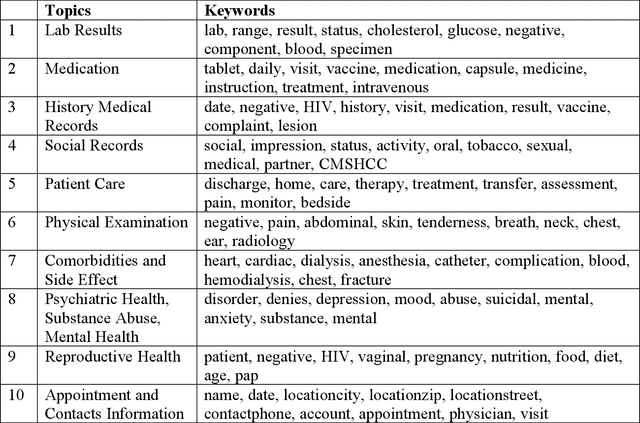
Objective: To characterize stigma dimensions, social, and related behavioral circumstances in people living with HIV (PLWHs) seeking care, using natural language processing methods applied to a large collection of electronic health record (EHR) clinical notes from a large integrated health system in the southeast United States. Methods: We identified 9,140 cohort of PLWHs from the UF Health IDR and performed topic modeling analysis using Latent Dirichlet Allocation (LDA) to uncover stigma dimensions, social, and related behavioral circumstances. Domain experts created a seed list of HIV-related stigma keywords, then applied a snowball strategy to iteratively review notes for additional terms until saturation was reached. To identify more target topics, we tested three keyword-based filtering strategies. Domain experts manually reviewed the detected topics using the prevalent terms and key discussion topics. Word frequency analysis was used to highlight the prevalent terms associated with each topic. In addition, we conducted topic variation analysis among subgroups to examine differences across age and sex-specific demographics. Results and Conclusion: Topic modeling on sentences containing at least one keyword uncovered a wide range of topic themes associated with HIV-related stigma, social, and related behaviors circumstances, including "Mental Health Concern and Stigma", "Social Support and Engagement", "Limited Healthcare Access and Severe Illness", "Treatment Refusal and Isolation" and so on. Topic variation analysis across age subgroups revealed differences. Extracting and understanding the HIV-related stigma dimensions, social, and related behavioral circumstances from EHR clinical notes enables scalable, time-efficient assessment, overcoming the limitations of traditional questionnaires and improving patient outcomes.
Identifying Symptoms of Delirium from Clinical Narratives Using Natural Language Processing
Mar 31, 2023



Delirium is an acute decline or fluctuation in attention, awareness, or other cognitive function that can lead to serious adverse outcomes. Despite the severe outcomes, delirium is frequently unrecognized and uncoded in patients' electronic health records (EHRs) due to its transient and diverse nature. Natural language processing (NLP), a key technology that extracts medical concepts from clinical narratives, has shown great potential in studies of delirium outcomes and symptoms. To assist in the diagnosis and phenotyping of delirium, we formed an expert panel to categorize diverse delirium symptoms, composed annotation guidelines, created a delirium corpus with diverse delirium symptoms, and developed NLP methods to extract delirium symptoms from clinical notes. We compared 5 state-of-the-art transformer models including 2 models (BERT and RoBERTa) from the general domain and 3 models (BERT_MIMIC, RoBERTa_MIMIC, and GatorTron) from the clinical domain. GatorTron achieved the best strict and lenient F1 scores of 0.8055 and 0.8759, respectively. We conducted an error analysis to identify challenges in annotating delirium symptoms and developing NLP systems. To the best of our knowledge, this is the first large language model-based delirium symptom extraction system. Our study lays the foundation for the future development of computable phenotypes and diagnosis methods for delirium.
DR-VIDAL -- Doubly Robust Variational Information-theoretic Deep Adversarial Learning for Counterfactual Prediction and Treatment Effect Estimation on Real World Data
Mar 07, 2023Determining causal effects of interventions onto outcomes from real-world, observational (non-randomized) data, e.g., treatment repurposing using electronic health records, is challenging due to underlying bias. Causal deep learning has improved over traditional techniques for estimating individualized treatment effects (ITE). We present the Doubly Robust Variational Information-theoretic Deep Adversarial Learning (DR-VIDAL), a novel generative framework that combines two joint models of treatment and outcome, ensuring an unbiased ITE estimation even when one of the two is misspecified. DR-VIDAL integrates: (i) a variational autoencoder (VAE) to factorize confounders into latent variables according to causal assumptions; (ii) an information-theoretic generative adversarial network (Info-GAN) to generate counterfactuals; (iii) a doubly robust block incorporating treatment propensities for outcome predictions. On synthetic and real-world datasets (Infant Health and Development Program, Twin Birth Registry, and National Supported Work Program), DR-VIDAL achieves better performance than other non-generative and generative methods. In conclusion, DR-VIDAL uniquely fuses causal assumptions, VAE, Info-GAN, and doubly robustness into a comprehensive, performant framework. Code is available at: https://github.com/Shantanu48114860/DR-VIDAL-AMIA-22 under MIT license.
Variational Temporal Deconfounder for Individualized Treatment Effect Estimation from Longitudinal Observational Data
Jul 23, 2022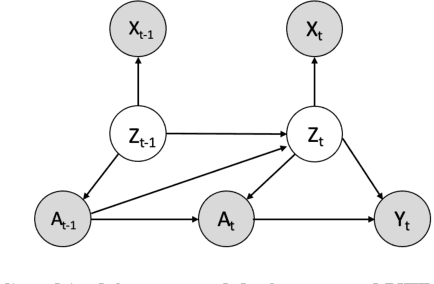
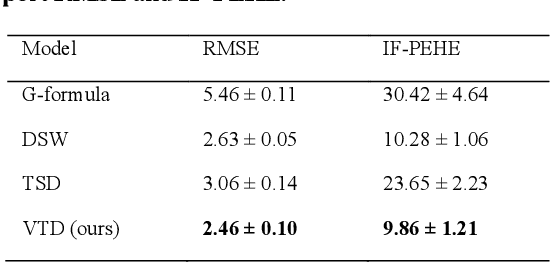
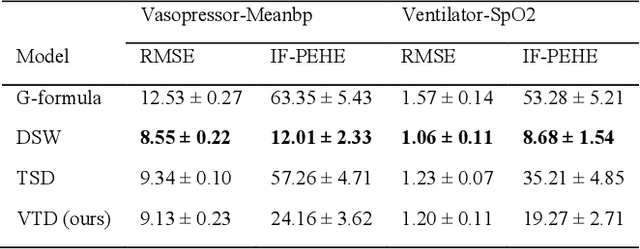
Estimating treatment effects, especially individualized treatment effects (ITE), using observational data is challenging due to the complex situations of confounding bias. Existing approaches for estimating treatment effects from longitudinal observational data are usually built upon a strong assumption of "unconfoundedness", which is hard to fulfill in real-world practice. In this paper, we propose the Variational Temporal Deconfounder (VTD), an approach that leverages deep variational embeddings in the longitudinal setting using proxies (i.e., surrogate variables that serve for unobservable variables). Specifically, VTD leverages observed proxies to learn a hidden embedding that reflects the true hidden confounders in the observational data. As such, our VTD method does not rely on the "unconfoundedness" assumption. We test our VTD method on both synthetic and real-world clinical data, and the results show that our approach is effective when hidden confounding is the leading bias compared to other existing models.
Joint Application of the Target Trial Causal Framework and Machine Learning Modeling to Optimize Antibiotic Therapy: Use Case on Acute Bacterial Skin and Skin Structure Infections due to Methicillin-resistant Staphylococcus aureus
Jul 15, 2022
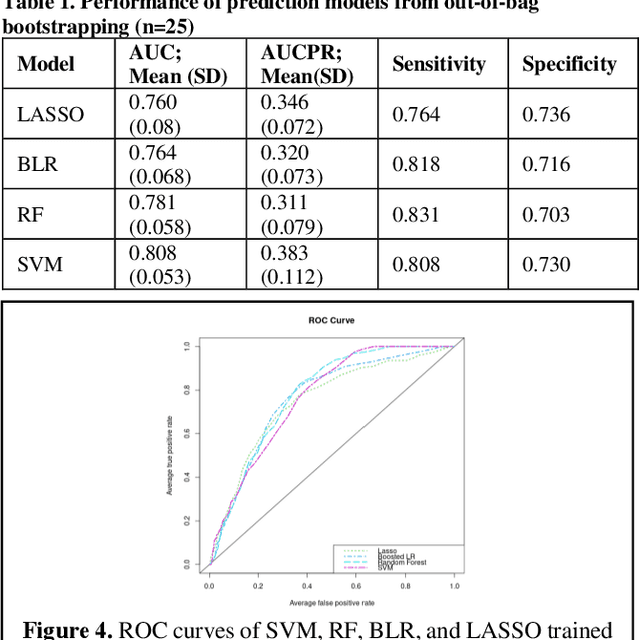
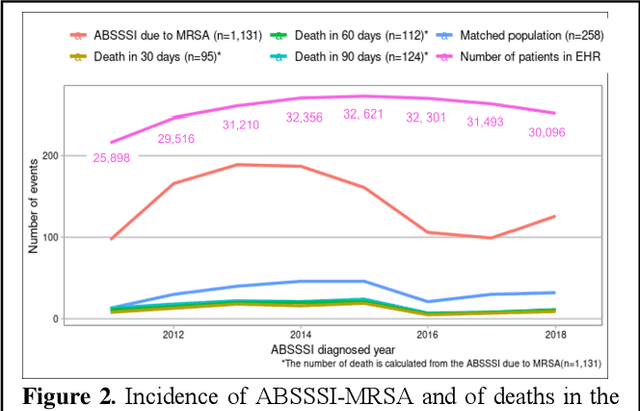
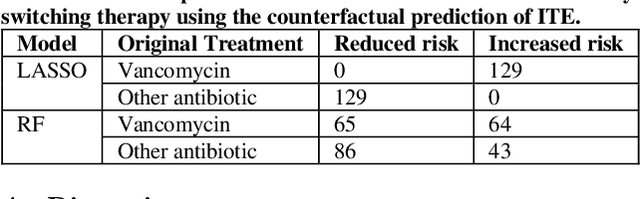
Bacterial infections are responsible for high mortality worldwide. Antimicrobial resistance underlying the infection, and multifaceted patient's clinical status can hamper the correct choice of antibiotic treatment. Randomized clinical trials provide average treatment effect estimates but are not ideal for risk stratification and optimization of therapeutic choice, i.e., individualized treatment effects (ITE). Here, we leverage large-scale electronic health record data, collected from Southern US academic clinics, to emulate a clinical trial, i.e., 'target trial', and develop a machine learning model of mortality prediction and ITE estimation for patients diagnosed with acute bacterial skin and skin structure infection (ABSSSI) due to methicillin-resistant Staphylococcus aureus (MRSA). ABSSSI-MRSA is a challenging condition with reduced treatment options - vancomycin is the preferred choice, but it has non-negligible side effects. First, we use propensity score matching to emulate the trial and create a treatment randomized (vancomycin vs. other antibiotics) dataset. Next, we use this data to train various machine learning methods (including boosted/LASSO logistic regression, support vector machines, and random forest) and choose the best model in terms of area under the receiver characteristic (AUC) through bootstrap validation. Lastly, we use the models to calculate ITE and identify possible averted deaths by therapy change. The out-of-bag tests indicate that SVM and RF are the most accurate, with AUC of 81% and 78%, respectively, but BLR/LASSO is not far behind (76%). By calculating the counterfactuals using the BLR/LASSO, vancomycin increases the risk of death, but it shows a large variation (odds ratio 1.2, 95% range 0.4-3.8) and the contribution to outcome probability is modest. Instead, the RF exhibits stronger changes in ITE, suggesting more complex treatment heterogeneity.
Assessing putative bias in prediction of anti-microbial resistance from real-world genotyping data under explicit causal assumptions
Jul 23, 2021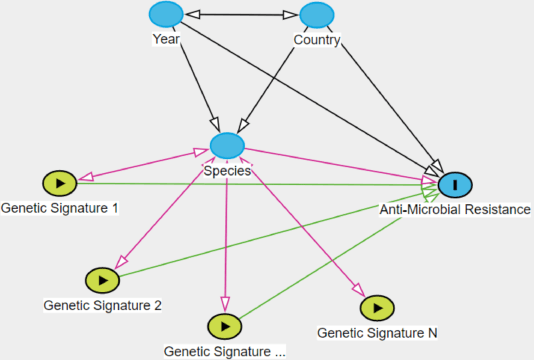
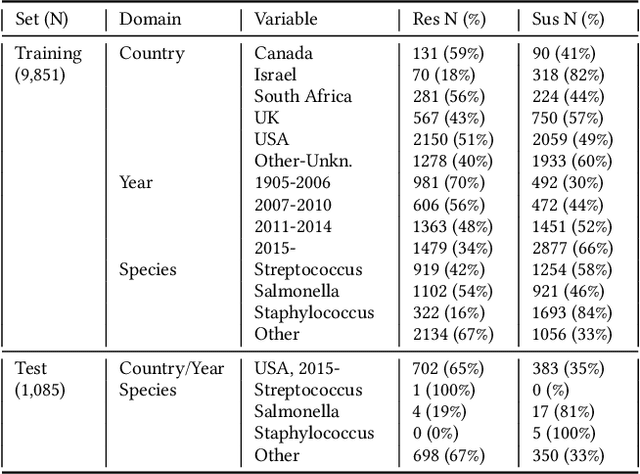
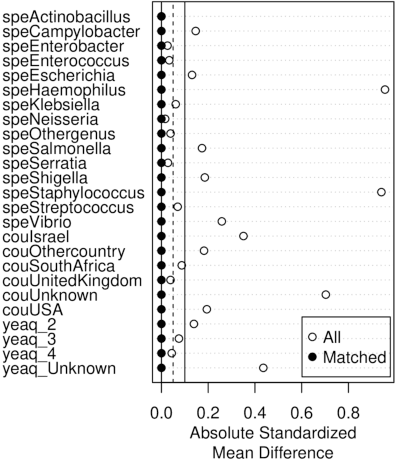
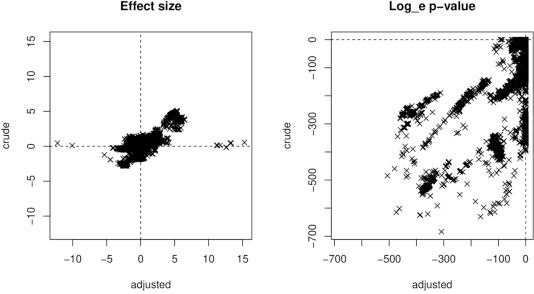
Whole genome sequencing (WGS) is quickly becoming the customary means for identification of antimicrobial resistance (AMR) due to its ability to obtain high resolution information about the genes and mechanisms that are causing resistance and driving pathogen mobility. By contrast, traditional phenotypic (antibiogram) testing cannot easily elucidate such information. Yet development of AMR prediction tools from genotype-phenotype data can be biased, since sampling is non-randomized. Sample provenience, period of collection, and species representation can confound the association of genetic traits with AMR. Thus, prediction models can perform poorly on new data with sampling distribution shifts. In this work -- under an explicit set of causal assumptions -- we evaluate the effectiveness of propensity-based rebalancing and confounding adjustment on AMR prediction using genotype-phenotype AMR data from the Pathosystems Resource Integration Center (PATRIC). We select bacterial genotypes (encoded as k-mer signatures, i.e. DNA fragments of length k), country, year, species, and AMR phenotypes for the tetracycline drug class, preparing test data with recent genomes coming from a single country. We test boosted logistic regression (BLR) and random forests (RF) with/without bias-handling. On 10,936 instances, we find evidence of species, location and year imbalance with respect to the AMR phenotype. The crude versus bias-adjusted change in effect of genetic signatures on AMR varies but only moderately (selecting the top 20,000 out of 40+ million k-mers). The area under the receiver operating characteristic (AUROC) of the RF (0.95) is comparable to that of BLR (0.94) on both out-of-bag samples from bootstrap and the external test (n=1,085), where AUROCs do not decrease. We observe a 1%-5% gain in AUROC with bias-handling compared to the sole use of genetic signatures. ...
Integrating Crowdsourcing and Active Learning for Classification of Work-Life Events from Tweets
Apr 02, 2020



Social media, especially Twitter, is being increasingly used for research with predictive analytics. In social media studies, natural language processing (NLP) techniques are used in conjunction with expert-based, manual and qualitative analyses. However, social media data are unstructured and must undergo complex manipulation for research use. The manual annotation is the most resource and time-consuming process that multiple expert raters have to reach consensus on every item, but is essential to create gold-standard datasets for training NLP-based machine learning classifiers. To reduce the burden of the manual annotation, yet maintaining its reliability, we devised a crowdsourcing pipeline combined with active learning strategies. We demonstrated its effectiveness through a case study that identifies job loss events from individual tweets. We used Amazon Mechanical Turk platform to recruit annotators from the Internet and designed a number of quality control measures to assure annotation accuracy. We evaluated 4 different active learning strategies (i.e., least confident, entropy, vote entropy, and Kullback-Leibler divergence). The active learning strategies aim at reducing the number of tweets needed to reach a desired performance of automated classification. Results show that crowdsourcing is useful to create high-quality annotations and active learning helps in reducing the number of required tweets, although there was no substantial difference among the strategies tested.
Mining Twitter to Assess the Determinants of Health Behavior towards Human Papillomavirus Vaccination in the United States
Jul 06, 2019

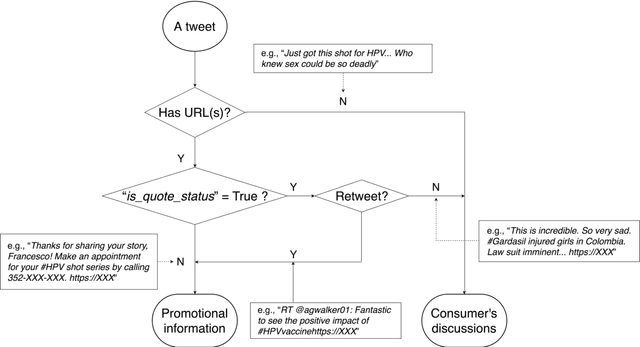

Objectives To test the feasibility of using Twitter data to assess determinants of consumers' health behavior towards Human papillomavirus (HPV) vaccination informed by the Integrated Behavior Model (IBM). Methods We used three Twitter datasets spanning from 2014 to 2018. We preprocessed and geocoded the tweets, and then built a rule-based model that classified each tweet into either promotional information or consumers' discussions. We applied topic modeling to discover major themes, and subsequently explored the associations between the topics learned from consumers' discussions and the responses of HPV-related questions in the Health Information National Trends Survey (HINTS). Results We collected 2,846,495 tweets and analyzed 335,681 geocoded tweets. Through topic modeling, we identified 122 high-quality topics. The most discussed consumer topic is "cervical cancer screening"; while in promotional tweets, the most popular topic is to increase awareness of "HPV causes cancer". 87 out of the 122 topics are correlated between promotional information and consumers' discussions. Guided by IBM, we examined the alignment between our Twitter findings and the results obtained from HINTS. 35 topics can be mapped to HINTS questions by keywords, 112 topics can be mapped to IBM constructs, and 45 topics have statistically significant correlations with HINTS responses in terms of geographic distributions. Conclusion Not only mining Twitter to assess consumers' health behaviors can obtain results comparable to surveys but can yield additional insights via a theory-driven approach. Limitations exist, nevertheless, these encouraging results impel us to develop innovative ways of leveraging social media in the changing health communication landscape.
Understanding Perceptions and Attitudes in Breast Cancer Discussions on Twitter
May 22, 2019
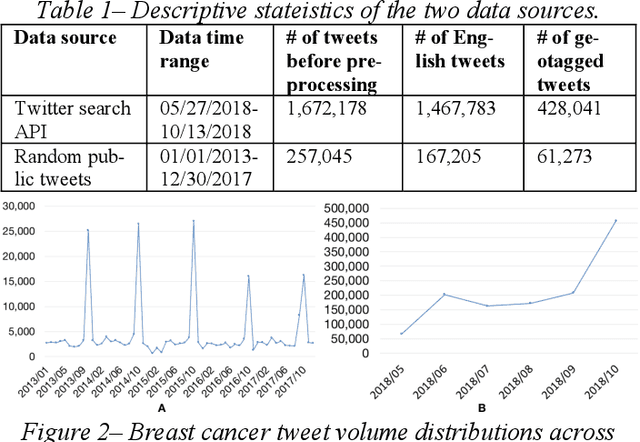
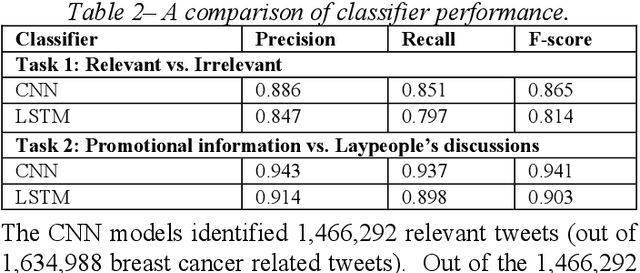
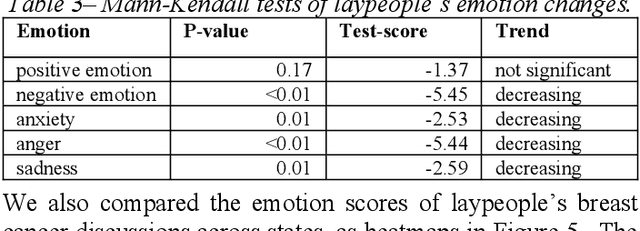
Among American women, the rate of breast cancer is only second to lung cancer. An estimated 12.4% women will develop breast cancer over the course of their lifetime. The widespread use of social media across the socio-economic spectrum offers unparalleled ways to facilitate information sharing, in particular as it pertains to health. Social media is also used by many healthcare stakeholders, ranging from government agencies to healthcare industry, to disseminate health information and to engage patients. The purpose of this study is to investigate people's perceptions and attitudes relate to breast cancer, especially those that are related to physical activities, on Twitter. To achieve this, we first identified and collected tweets related to breast cancer; and then used topic modeling and sentiment analysis techniques to understanding discussion themes and quantify Twitter users' perceptions and emotions w.r.t breast cancer to answer 5 research questions.
Inference of a Multi-Domain Machine Learning Model to Predict Mortality in Hospital Stays for Patients with Cancer upon Febrile Neutropenia Onset
Feb 27, 2019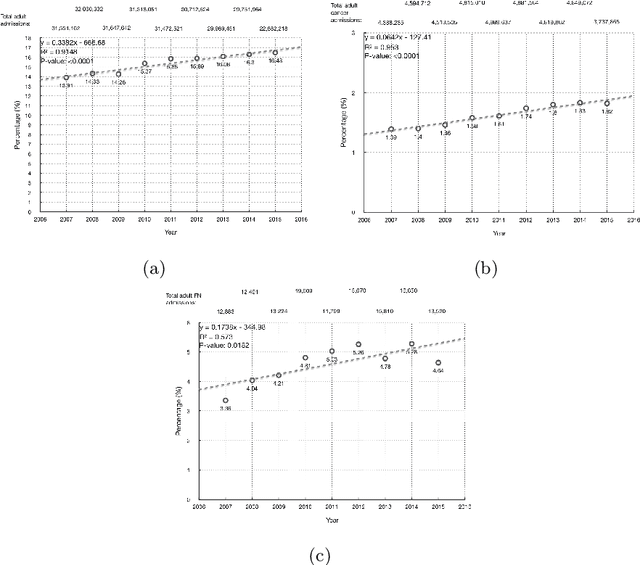

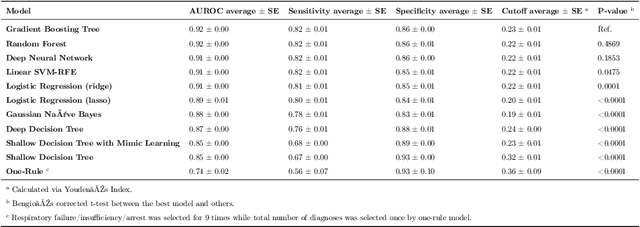
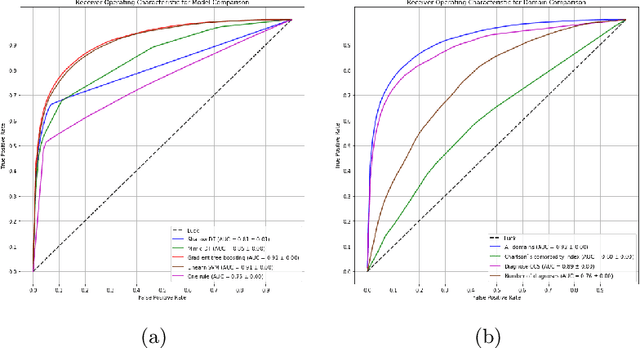
Febrile neutropenia (FN) has been associated with high mortality, especially among adults with cancer. Understanding the patient and provider level heterogeneity in FN hospital admissions has potential to inform personalized interventions focused on increasing survival of individuals with FN. We leverage machine learning techniques to disentangling the complex interactions among multi domain risk factors in a population with FN. Data from the Healthcare Cost and Utilization Project (HCUP) National Inpatient Sample and Nationwide Inpatient Sample (NIS) were used to build machine learning based models of mortality for adult cancer patients who were diagnosed with FN during a hospital admission. In particular, the importance of risk factors from different domains (including demographic, clinical, and hospital associated information) was studied. A set of more interpretable (decision tree, logistic regression) as well as more black box (random forest, gradient boosting, neural networks) models were analyzed and compared via multiple cross validation. Our results demonstrate that a linear prediction score of FN mortality among adults with cancer, based on admission information is effective in classifying high risk patients; clinical diagnoses is the domain with the highest predictive power. A number of the risk variables (e.g. sepsis, kidney failure, etc.) identified in this study are clinically actionable and may inform future studies looking at the patients prior medical history are warranted.