 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeJoint Application of the Target Trial Causal Framework and Machine Learning Modeling to Optimize Antibiotic Therapy: Use Case on Acute Bacterial Skin and Skin Structure Infections due to Methicillin-resistant Staphylococcus aureus
Jul 15, 2022
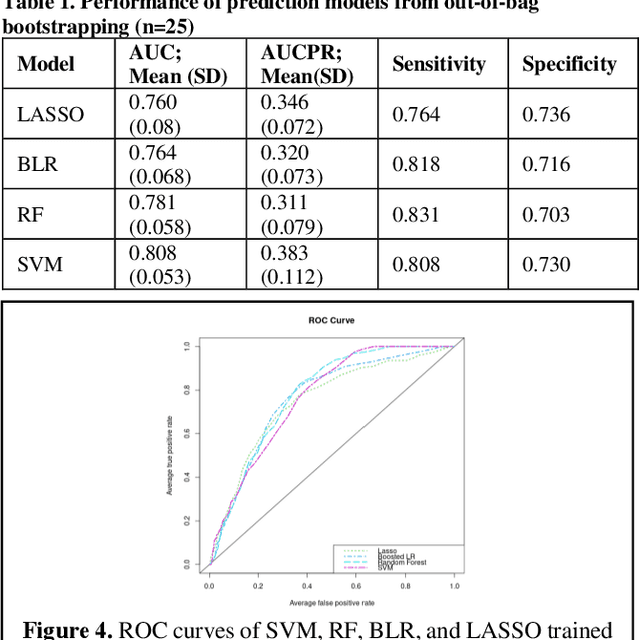
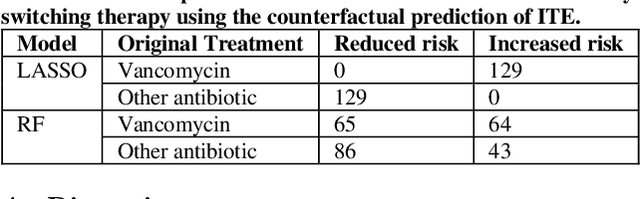
Bacterial infections are responsible for high mortality worldwide. Antimicrobial resistance underlying the infection, and multifaceted patient's clinical status can hamper the correct choice of antibiotic treatment. Randomized clinical trials provide average treatment effect estimates but are not ideal for risk stratification and optimization of therapeutic choice, i.e., individualized treatment effects (ITE). Here, we leverage large-scale electronic health record data, collected from Southern US academic clinics, to emulate a clinical trial, i.e., 'target trial', and develop a machine learning model of mortality prediction and ITE estimation for patients diagnosed with acute bacterial skin and skin structure infection (ABSSSI) due to methicillin-resistant Staphylococcus aureus (MRSA). ABSSSI-MRSA is a challenging condition with reduced treatment options - vancomycin is the preferred choice, but it has non-negligible side effects. First, we use propensity score matching to emulate the trial and create a treatment randomized (vancomycin vs. other antibiotics) dataset. Next, we use this data to train various machine learning methods (including boosted/LASSO logistic regression, support vector machines, and random forest) and choose the best model in terms of area under the receiver characteristic (AUC) through bootstrap validation. Lastly, we use the models to calculate ITE and identify possible averted deaths by therapy change. The out-of-bag tests indicate that SVM and RF are the most accurate, with AUC of 81% and 78%, respectively, but BLR/LASSO is not far behind (76%). By calculating the counterfactuals using the BLR/LASSO, vancomycin increases the risk of death, but it shows a large variation (odds ratio 1.2, 95% range 0.4-3.8) and the contribution to outcome probability is modest. Instead, the RF exhibits stronger changes in ITE, suggesting more complex treatment heterogeneity.
Assessing putative bias in prediction of anti-microbial resistance from real-world genotyping data under explicit causal assumptions
Jul 23, 2021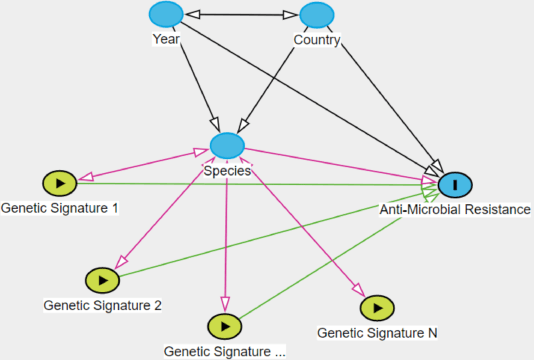
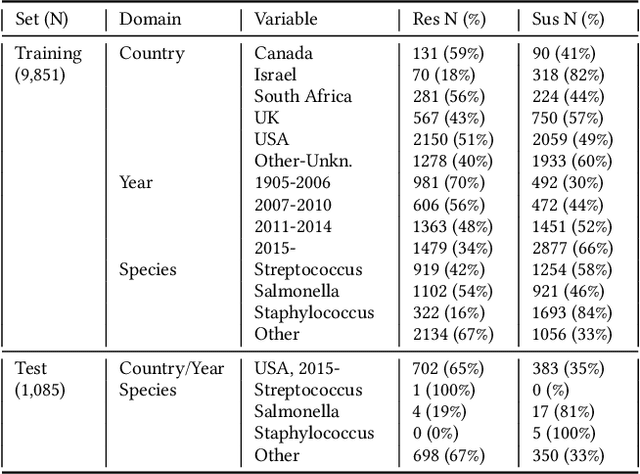
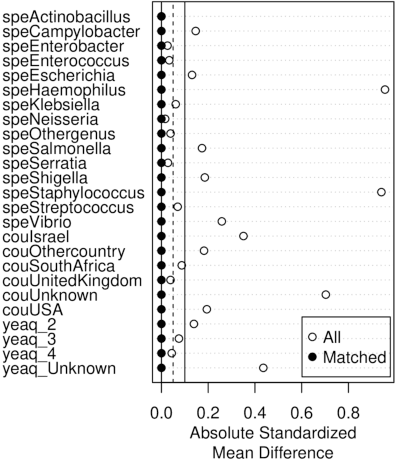
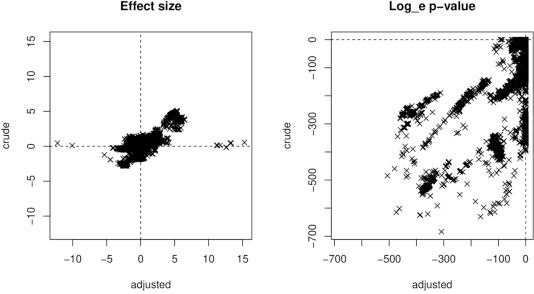
Whole genome sequencing (WGS) is quickly becoming the customary means for identification of antimicrobial resistance (AMR) due to its ability to obtain high resolution information about the genes and mechanisms that are causing resistance and driving pathogen mobility. By contrast, traditional phenotypic (antibiogram) testing cannot easily elucidate such information. Yet development of AMR prediction tools from genotype-phenotype data can be biased, since sampling is non-randomized. Sample provenience, period of collection, and species representation can confound the association of genetic traits with AMR. Thus, prediction models can perform poorly on new data with sampling distribution shifts. In this work -- under an explicit set of causal assumptions -- we evaluate the effectiveness of propensity-based rebalancing and confounding adjustment on AMR prediction using genotype-phenotype AMR data from the Pathosystems Resource Integration Center (PATRIC). We select bacterial genotypes (encoded as k-mer signatures, i.e. DNA fragments of length k), country, year, species, and AMR phenotypes for the tetracycline drug class, preparing test data with recent genomes coming from a single country. We test boosted logistic regression (BLR) and random forests (RF) with/without bias-handling. On 10,936 instances, we find evidence of species, location and year imbalance with respect to the AMR phenotype. The crude versus bias-adjusted change in effect of genetic signatures on AMR varies but only moderately (selecting the top 20,000 out of 40+ million k-mers). The area under the receiver operating characteristic (AUROC) of the RF (0.95) is comparable to that of BLR (0.94) on both out-of-bag samples from bootstrap and the external test (n=1,085), where AUROCs do not decrease. We observe a 1%-5% gain in AUROC with bias-handling compared to the sole use of genetic signatures. ...
Underwater Fish Detection with Weak Multi-Domain Supervision
May 26, 2019



Given a sufficiently large training dataset, it is relatively easy to train a modern convolution neural network (CNN) as a required image classifier. However, for the task of fish classification and/or fish detection, if a CNN was trained to detect or classify particular fish species in particular background habitats, the same CNN exhibits much lower accuracy when applied to new/unseen fish species and/or fish habitats. Therefore, in practice, the CNN needs to be continuously fine-tuned to improve its classification accuracy to handle new project-specific fish species or habitats. In this work we present a labelling-efficient method of training a CNN-based fish-detector (the Xception CNN was used as the base) on relatively small numbers (4,000) of project-domain underwater fish/no-fish images from 20 different habitats. Additionally, 17,000 of known negative (that is, missing fish) general-domain (VOC2012) above-water images were used. Two publicly available fish-domain datasets supplied additional 27,000 of above-water and underwater positive/fish images. By using this multi-domain collection of images, the trained Xception-based binary (fish/not-fish) classifier achieved 0.17% false-positives and 0.61% false-negatives on the project's 20,000 negative and 16,000 positive holdout test images, respectively. The area under the ROC curve (AUC) was 99.94%.