 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAssessing putative bias in prediction of anti-microbial resistance from real-world genotyping data under explicit causal assumptions
Jul 23, 2021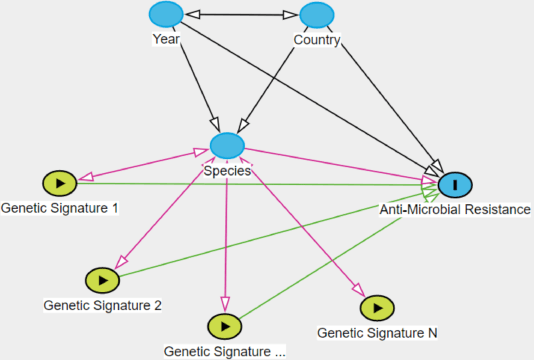
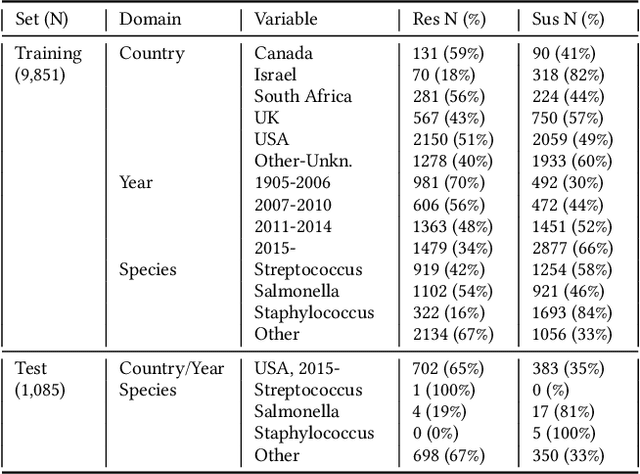
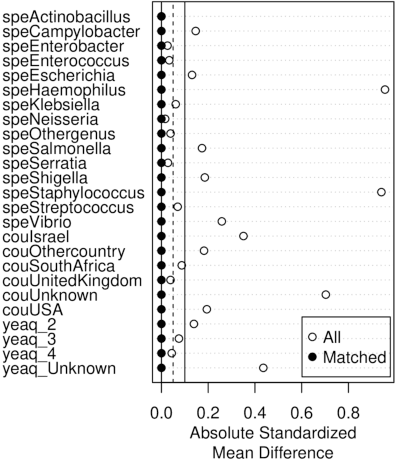
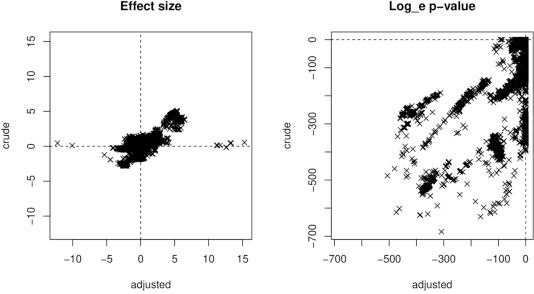
Whole genome sequencing (WGS) is quickly becoming the customary means for identification of antimicrobial resistance (AMR) due to its ability to obtain high resolution information about the genes and mechanisms that are causing resistance and driving pathogen mobility. By contrast, traditional phenotypic (antibiogram) testing cannot easily elucidate such information. Yet development of AMR prediction tools from genotype-phenotype data can be biased, since sampling is non-randomized. Sample provenience, period of collection, and species representation can confound the association of genetic traits with AMR. Thus, prediction models can perform poorly on new data with sampling distribution shifts. In this work -- under an explicit set of causal assumptions -- we evaluate the effectiveness of propensity-based rebalancing and confounding adjustment on AMR prediction using genotype-phenotype AMR data from the Pathosystems Resource Integration Center (PATRIC). We select bacterial genotypes (encoded as k-mer signatures, i.e. DNA fragments of length k), country, year, species, and AMR phenotypes for the tetracycline drug class, preparing test data with recent genomes coming from a single country. We test boosted logistic regression (BLR) and random forests (RF) with/without bias-handling. On 10,936 instances, we find evidence of species, location and year imbalance with respect to the AMR phenotype. The crude versus bias-adjusted change in effect of genetic signatures on AMR varies but only moderately (selecting the top 20,000 out of 40+ million k-mers). The area under the receiver operating characteristic (AUROC) of the RF (0.95) is comparable to that of BLR (0.94) on both out-of-bag samples from bootstrap and the external test (n=1,085), where AUROCs do not decrease. We observe a 1%-5% gain in AUROC with bias-handling compared to the sole use of genetic signatures. ...