 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Multi-Agent System for Information Extraction from the Chemical Literature
Jul 27, 2025To fully expedite AI-powered chemical research, high-quality chemical databases are the cornerstone. Automatic extraction of chemical information from the literature is essential for constructing reaction databases, but it is currently limited by the multimodality and style variability of chemical information. In this work, we developed a multimodal large language model (MLLM)-based multi-agent system for automatic chemical information extraction. We used the MLLM's strong reasoning capability to understand the structure of complex chemical graphics, decompose the extraction task into sub-tasks and coordinate a set of specialized agents to solve them. Our system achieved an F1 score of 80.8% on a benchmark dataset of complex chemical reaction graphics from the literature, surpassing the previous state-of-the-art model (F1 score: 35.6%) by a significant margin. Additionally, it demonstrated consistent improvements in key sub-tasks, including molecular image recognition, reaction image parsing, named entity recognition and text-based reaction extraction. This work is a critical step toward automated chemical information extraction into structured datasets, which will be a strong promoter of AI-driven chemical research.
Towards Large-scale Chemical Reaction Image Parsing via a Multimodal Large Language Model
Mar 11, 2025Artificial intelligence (AI) has demonstrated significant promise in advancing organic chemistry research; however, its effectiveness depends on the availability of high-quality chemical reaction data. Currently, most published chemical reactions are not available in machine-readable form, limiting the broader application of AI in this field. The extraction of published chemical reactions into structured databases still relies heavily on manual curation, and robust automatic parsing of chemical reaction images into machine-readable data remains a significant challenge. To address this, we introduce the Reaction Image Multimodal large language model (RxnIM), the first multimodal large language model specifically designed to parse chemical reaction images into machine-readable reaction data. RxnIM not only extracts key chemical components from reaction images but also interprets the textual content that describes reaction conditions. Together with specially designed large-scale dataset generation method to support model training, our approach achieves excellent performance, with an average F1 score of 88% on various benchmarks, surpassing literature methods by 5%. This represents a crucial step toward the automatic construction of large databases of machine-readable reaction data parsed from images in the chemistry literature, providing essential data resources for AI research in chemistry. The source code, model checkpoints, and datasets developed in this work are released under permissive licenses. An instance of the RxnIM web application can be accessed at https://huggingface.co/spaces/CYF200127/RxnIM.
MG-3D: Multi-Grained Knowledge-Enhanced 3D Medical Vision-Language Pre-training
Dec 08, 2024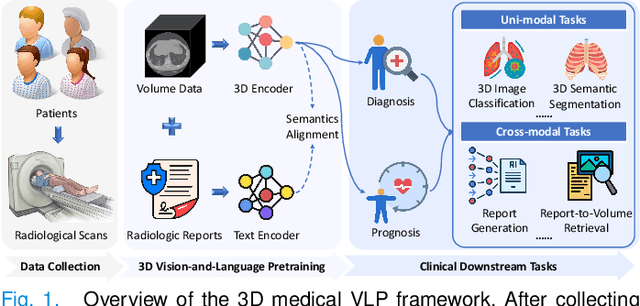

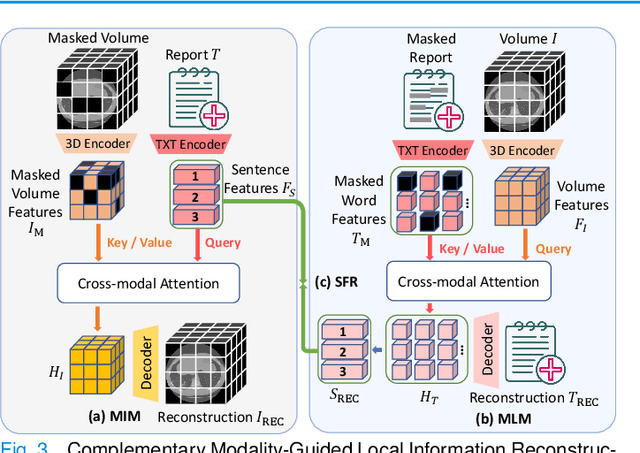

3D medical image analysis is pivotal in numerous clinical applications. However, the scarcity of labeled data and limited generalization capabilities hinder the advancement of AI-empowered models. Radiology reports are easily accessible and can serve as weakly-supervised signals. However, large-scale vision-language pre-training (VLP) remains underexplored in 3D medical image analysis. Specifically, the insufficient investigation into multi-grained radiology semantics and their correlations across patients leads to underutilization of large-scale volume-report data. Considering intra-patient cross-modal semantic consistency and inter-patient semantic correlations, we propose a multi-task VLP method, MG-3D, pre-trained on large-scale data (47.1K), addressing the challenges by the following two aspects: 1) Establishing the correspondence between volume semantics and multi-grained medical knowledge of each patient with cross-modal global alignment and complementary modality-guided local reconstruction, ensuring intra-patient features of different modalities cohesively represent the same semantic content; 2) Correlating inter-patient visual semantics based on fine-grained report correlations across patients, and keeping sensitivity to global individual differences via contrastive learning, enhancing the discriminative feature representation. Furthermore, we delve into the scaling law to explore potential performance improvements. Comprehensive evaluations across nine uni- and cross-modal clinical tasks are carried out to assess model efficacy. Extensive experiments on both internal and external datasets demonstrate the superior transferability, scalability, and generalization of MG-3D, showcasing its potential in advancing feature representation for 3D medical image analysis. Code will be available: https://github.com/Xuefeng-Ni/MG-3D.
SMiCRM: A Benchmark Dataset of Mechanistic Molecular Images
Jul 25, 2024
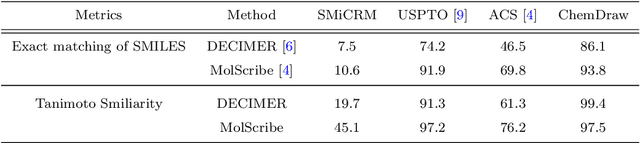
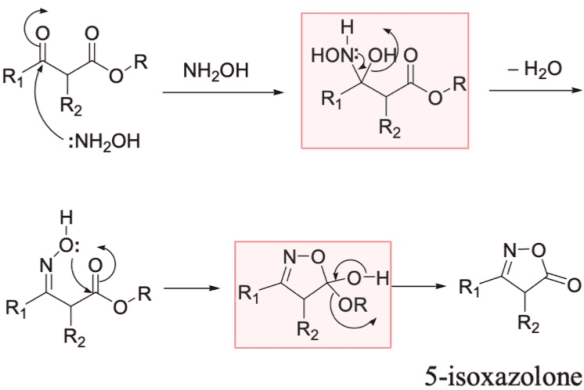
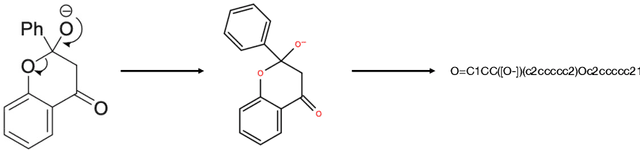
Optical chemical structure recognition (OCSR) systems aim to extract the molecular structure information, usually in the form of molecular graph or SMILES, from images of chemical molecules. While many tools have been developed for this purpose, challenges still exist due to different types of noises that might exist in the images. Specifically, we focus on the 'arrow-pushing' diagrams, a typical type of chemical images to demonstrate electron flow in mechanistic steps. We present Structural molecular identifier of Molecular images in Chemical Reaction Mechanisms (SMiCRM), a dataset designed to benchmark machine recognition capabilities of chemical molecules with arrow-pushing annotations. Comprising 453 images, it spans a broad array of organic chemical reactions, each illustrated with molecular structures and mechanistic arrows. SMiCRM offers a rich collection of annotated molecule images for enhancing the benchmarking process for OCSR methods. This dataset includes a machine-readable molecular identity for each image as well as mechanistic arrows showing electron flow during chemical reactions. It presents a more authentic and challenging task for testing molecular recognition technologies, and achieving this task can greatly enrich the mechanisitic information in computer-extracted chemical reaction data.
MolNexTR: A Generalized Deep Learning Model for Molecular Image Recognition
Mar 08, 2024

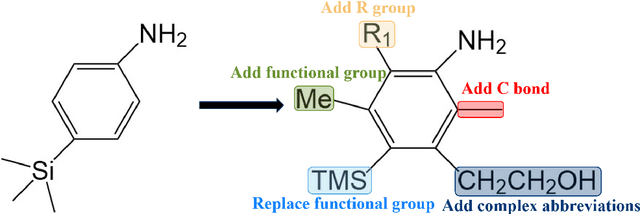
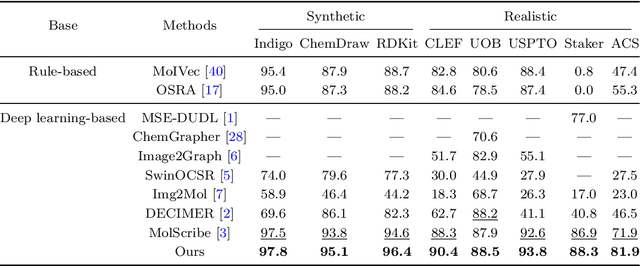
In the field of chemical structure recognition, the task of converting molecular images into graph structures and SMILES string stands as a significant challenge, primarily due to the varied drawing styles and conventions prevalent in chemical literature. To bridge this gap, we proposed MolNexTR, a novel image-to-graph deep learning model that collaborates to fuse the strengths of ConvNext, a powerful Convolutional Neural Network variant, and Vision-TRansformer. This integration facilitates a more nuanced extraction of both local and global features from molecular images. MolNexTR can predict atoms and bonds simultaneously and understand their layout rules. It also excels at flexibly integrating symbolic chemistry principles to discern chirality and decipher abbreviated structures. We further incorporate a series of advanced algorithms, including improved data augmentation module, image contamination module, and a post-processing module to get the final SMILES output. These modules synergistically enhance the model's robustness against the diverse styles of molecular imagery found in real literature. In our test sets, MolNexTR has demonstrated superior performance, achieving an accuracy rate of 81-97%, marking a significant advancement in the domain of molecular structure recognition. Scientific contribution: MolNexTR is a novel image-to-graph model that incorporates a unique dual-stream encoder to extract complex molecular image features, and combines chemical rules to predict atoms and bonds while understanding atom and bond layout rules. In addition, it employs a series of novel augmentation algorithms to significantly enhance the robustness and performance of the model.
RetroOOD: Understanding Out-of-Distribution Generalization in Retrosynthesis Prediction
Dec 18, 2023Machine learning-assisted retrosynthesis prediction models have been gaining widespread adoption, though their performances oftentimes degrade significantly when deployed in real-world applications embracing out-of-distribution (OOD) molecules or reactions. Despite steady progress on standard benchmarks, our understanding of existing retrosynthesis prediction models under the premise of distribution shifts remains stagnant. To this end, we first formally sort out two types of distribution shifts in retrosynthesis prediction and construct two groups of benchmark datasets. Next, through comprehensive experiments, we systematically compare state-of-the-art retrosynthesis prediction models on the two groups of benchmarks, revealing the limitations of previous in-distribution evaluation and re-examining the advantages of each model. More remarkably, we are motivated by the above empirical insights to propose two model-agnostic techniques that can improve the OOD generalization of arbitrary off-the-shelf retrosynthesis prediction algorithms. Our preliminary experiments show their high potential with an average performance improvement of 4.6%, and the established benchmarks serve as a foothold for further retrosynthesis prediction research towards OOD generalization.