 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeStrategic priorities for transformative progress in advancing biology with proteomics and artificial intelligence
Feb 21, 2025

Artificial intelligence (AI) is transforming scientific research, including proteomics. Advances in mass spectrometry (MS)-based proteomics data quality, diversity, and scale, combined with groundbreaking AI techniques, are unlocking new challenges and opportunities in biological discovery. Here, we highlight key areas where AI is driving innovation, from data analysis to new biological insights. These include developing an AI-friendly ecosystem for proteomics data generation, sharing, and analysis; improving peptide and protein identification and quantification; characterizing protein-protein interactions and protein complexes; advancing spatial and perturbation proteomics; integrating multi-omics data; and ultimately enabling AI-empowered virtual cells.
Contrastive Learning for Non-Local Graphs with Multi-Resolution Structural Views
Aug 19, 2023Learning node-level representations of heterophilic graphs is crucial for various applications, including fraudster detection and protein function prediction. In such graphs, nodes share structural similarity identified by the equivalence of their connectivity which is implicitly encoded in the form of higher-order hierarchical information in the graphs. The contrastive methods are popular choices for learning the representation of nodes in a graph. However, existing contrastive methods struggle to capture higher-order graph structures. To address this limitation, we propose a novel multiview contrastive learning approach that integrates diffusion filters on graphs. By incorporating multiple graph views as augmentations, our method captures the structural equivalence in heterophilic graphs, enabling the discovery of hidden relationships and similarities not apparent in traditional node representations. Our approach outperforms baselines on synthetic and real structural datasets, surpassing the best baseline by $16.06\%$ on Cornell, $3.27\%$ on Texas, and $8.04\%$ on Wisconsin. Additionally, it consistently achieves superior performance on proximal tasks, demonstrating its effectiveness in uncovering structural information and improving downstream applications.
Structured Q-learning For Antibody Design
Sep 13, 2022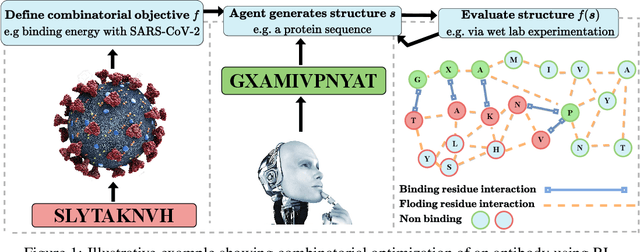
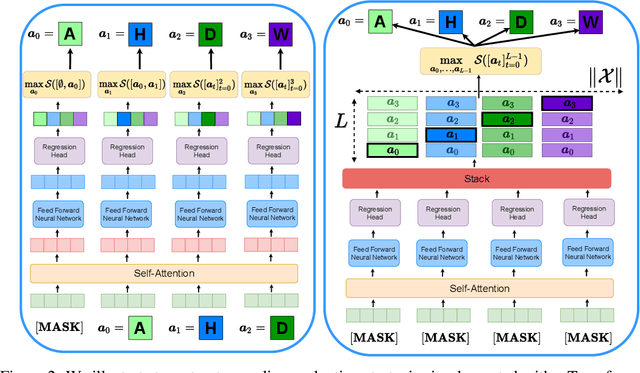
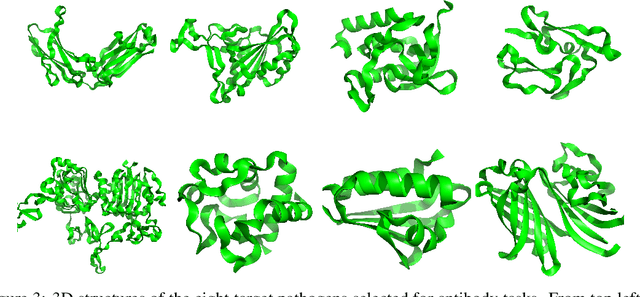
Optimizing combinatorial structures is core to many real-world problems, such as those encountered in life sciences. For example, one of the crucial steps involved in antibody design is to find an arrangement of amino acids in a protein sequence that improves its binding with a pathogen. Combinatorial optimization of antibodies is difficult due to extremely large search spaces and non-linear objectives. Even for modest antibody design problems, where proteins have a sequence length of eleven, we are faced with searching over 2.05 x 10^14 structures. Applying traditional Reinforcement Learning algorithms such as Q-learning to combinatorial optimization results in poor performance. We propose Structured Q-learning (SQL), an extension of Q-learning that incorporates structural priors for combinatorial optimization. Using a molecular docking simulator, we demonstrate that SQL finds high binding energy sequences and performs favourably against baselines on eight challenging antibody design tasks, including designing antibodies for SARS-COV.
Adversarial robustness of $β-$VAE through the lens of local geometry
Aug 08, 2022
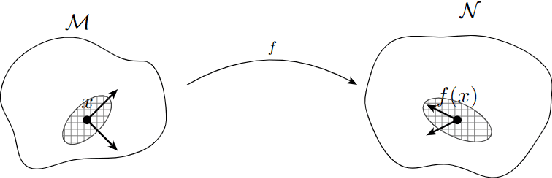
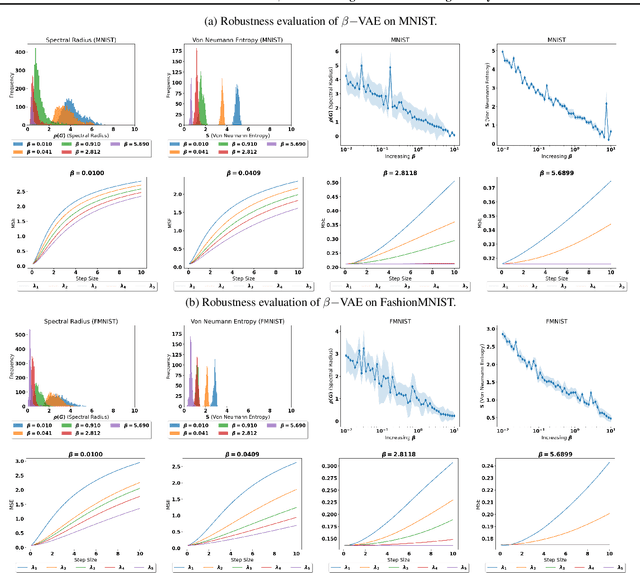
Variational autoencoders (VAEs) are susceptible to adversarial attacks. An adversary can find a small perturbation in the input sample to change its latent encoding non-smoothly, thereby compromising the reconstruction. A known reason for such vulnerability is the latent space distortions arising from a mismatch between approximated latent posterior and a prior distribution. Consequently, a slight change in the inputs leads to a significant change in the latent space encodings. This paper demonstrates that the sensitivity around a data point is due to a directional bias of a stochastic pullback metric tensor induced by the encoder network. The pullback metric tensor measures the infinitesimal volume change from input to latent space. Thus, it can be viewed as a lens to analyse the effect of small changes in the input leading to distortions in the latent space. We propose robustness evaluation scores using the eigenspectrum of a pullback metric. Moreover, we empirically show that the scores correlate with the robustness parameter $\beta$ of the $\beta-$VAE.
AntBO: Towards Real-World Automated Antibody Design with Combinatorial Bayesian Optimisation
Feb 16, 2022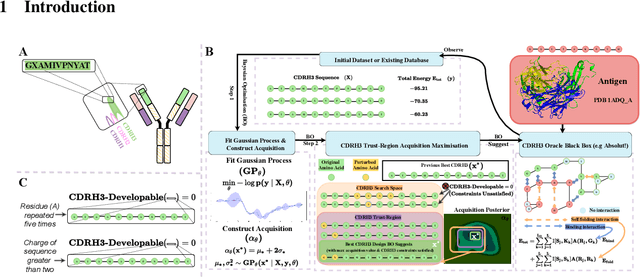
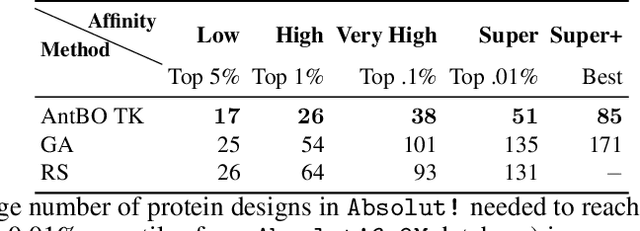
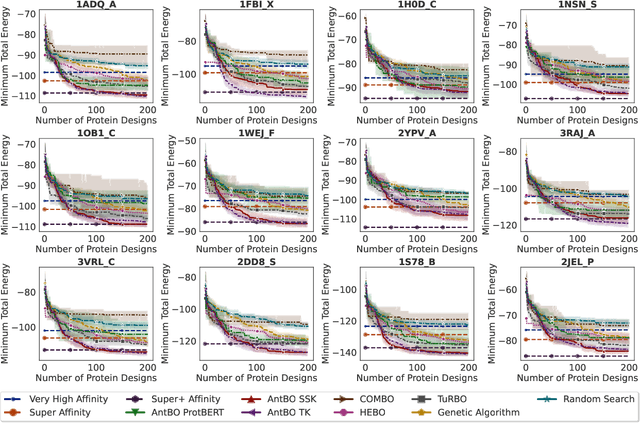
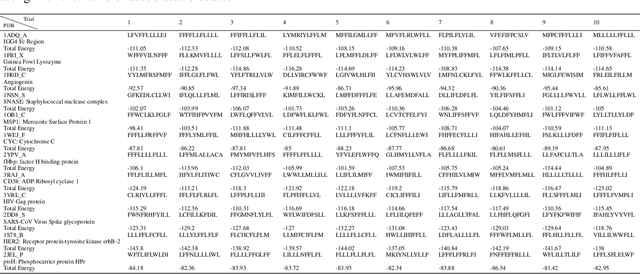
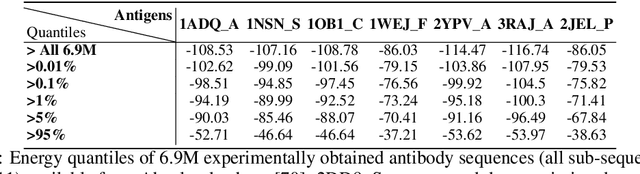
Antibodies are canonically Y-shaped multimeric proteins capable of highly specific molecular recognition. The CDRH3 region located at the tip of variable chains of an antibody dominates antigen-binding specificity. Therefore, it is a priority to design optimal antigen-specific CDRH3 regions to develop therapeutic antibodies to combat harmful pathogens. However, the combinatorial nature of CDRH3 sequence space makes it impossible to search for an optimal binding sequence exhaustively and efficiently, especially not experimentally. Here, we present AntBO: a Combinatorial Bayesian Optimisation framework enabling efficient in silico design of the CDRH3 region. Ideally, antibodies should bind to their target antigen and be free from any harmful outcomes. Therefore, we introduce the CDRH3 trust region that restricts the search to sequences with feasible developability scores. To benchmark AntBO, we use the Absolut! software suite as a black-box oracle because it can score the target specificity and affinity of designed antibodies in silico in an unconstrained fashion. The results across 188 antigens demonstrate the benefit of AntBO in designing CDRH3 regions with diverse biophysical properties. In under 200 protein designs, AntBO can suggest antibody sequences that outperform the best binding sequence drawn from 6.9 million experimentally obtained CDRH3s and a commonly used genetic algorithm baseline. Additionally, AntBO finds very-high affinity CDRH3 sequences in only 38 protein designs whilst requiring no domain knowledge. We conclude AntBO brings automated antibody design methods closer to what is practically viable for in vitro experimentation.
Hamiltonian prior to Disentangle Content and Motion in Image Sequences
Dec 02, 2021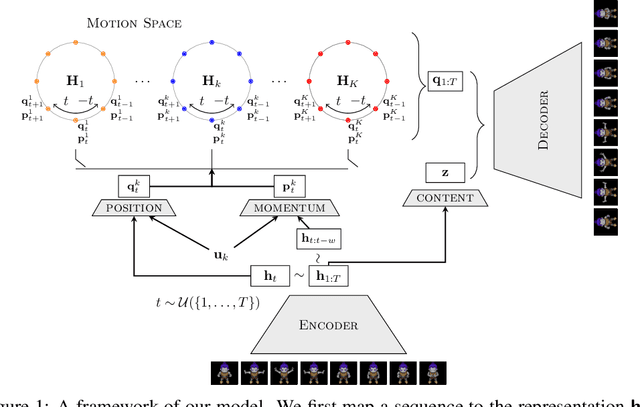
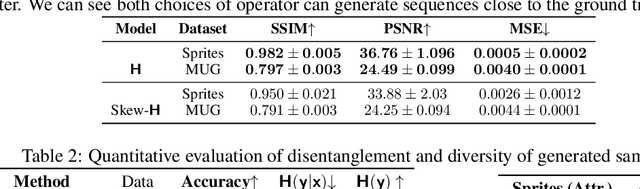


We present a deep latent variable model for high dimensional sequential data. Our model factorises the latent space into content and motion variables. To model the diverse dynamics, we split the motion space into subspaces, and introduce a unique Hamiltonian operator for each subspace. The Hamiltonian formulation provides reversible dynamics that learn to constrain the motion path to conserve invariant properties. The explicit split of the motion space decomposes the Hamiltonian into symmetry groups and gives long-term separability of the dynamics. This split also means representations can be learnt that are easy to interpret and control. We demonstrate the utility of our model for swapping the motion of two videos, generating sequences of various actions from a given image and unconditional sequence generation.