 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUnderstanding Task Vectors in In-Context Learning: Emergence, Functionality, and Limitations
Jun 10, 2025Task vectors offer a compelling mechanism for accelerating inference in in-context learning (ICL) by distilling task-specific information into a single, reusable representation. Despite their empirical success, the underlying principles governing their emergence and functionality remain unclear. This work proposes the Linear Combination Conjecture, positing that task vectors act as single in-context demonstrations formed through linear combinations of the original ones. We provide both theoretical and empirical support for this conjecture. First, we show that task vectors naturally emerge in linear transformers trained on triplet-formatted prompts through loss landscape analysis. Next, we predict the failure of task vectors on representing high-rank mappings and confirm this on practical LLMs. Our findings are further validated through saliency analyses and parameter visualization, suggesting an enhancement of task vectors by injecting multiple ones into few-shot prompts. Together, our results advance the understanding of task vectors and shed light on the mechanisms underlying ICL in transformer-based models.
AutoSDT: Scaling Data-Driven Discovery Tasks Toward Open Co-Scientists
Jun 09, 2025



Despite long-standing efforts in accelerating scientific discovery with AI, building AI co-scientists remains challenging due to limited high-quality data for training and evaluation. To tackle this data scarcity issue, we present AutoSDT, an automatic pipeline that collects high-quality coding tasks in real-world data-driven discovery workflows. AutoSDT leverages the coding capabilities and parametric knowledge of LLMs to search for diverse sources, select ecologically valid tasks, and synthesize accurate task instructions and code solutions. Using our pipeline, we construct AutoSDT-5K, a dataset of 5,404 coding tasks for data-driven discovery that covers four scientific disciplines and 756 unique Python packages. To the best of our knowledge, AutoSDT-5K is the only automatically collected and the largest open dataset for data-driven scientific discovery. Expert feedback on a subset of 256 tasks shows the effectiveness of AutoSDT: 93% of the collected tasks are ecologically valid, and 92.2% of the synthesized programs are functionally correct. Trained on AutoSDT-5K, the Qwen2.5-Coder-Instruct LLM series, dubbed AutoSDT-Coder, show substantial improvement on two challenging data-driven discovery benchmarks, ScienceAgentBench and DiscoveryBench. Most notably, AutoSDT-Coder-32B reaches the same level of performance as GPT-4o on ScienceAgentBench with a success rate of 7.8%, doubling the performance of its base model. On DiscoveryBench, it lifts the hypothesis matching score to 8.1, bringing a 17.4% relative improvement and closing the gap between open-weight models and GPT-4o.
Large Language Models for Controllable Multi-property Multi-objective Molecule Optimization
May 29, 2025In real-world drug design, molecule optimization requires selectively improving multiple molecular properties up to pharmaceutically relevant levels, while maintaining others that already meet such criteria. However, existing computational approaches and instruction-tuned LLMs fail to capture such nuanced property-specific objectives, limiting their practical applicability. To address this, we introduce C-MuMOInstruct, the first instruction-tuning dataset focused on multi-property optimization with explicit, property-specific objectives. Leveraging C-MuMOInstruct, we develop GeLLMO-Cs, a series of instruction-tuned LLMs that can perform targeted property-specific optimization. Our experiments across 5 in-distribution and 5 out-of-distribution tasks show that GeLLMO-Cs consistently outperform strong baselines, achieving up to 126% higher success rate. Notably, GeLLMO-Cs exhibit impressive 0-shot generalization to novel optimization tasks and unseen instructions. This offers a step toward a foundational LLM to support realistic, diverse optimizations with property-specific objectives. C-MuMOInstruct and code are accessible through https://github.com/ninglab/GeLLMO-C.
DiffER: Categorical Diffusion for Chemical Retrosynthesis
May 29, 2025Methods for automatic chemical retrosynthesis have found recent success through the application of models traditionally built for natural language processing, primarily through transformer neural networks. These models have demonstrated significant ability to translate between the SMILES encodings of chemical products and reactants, but are constrained as a result of their autoregressive nature. We propose DiffER, an alternative template-free method for retrosynthesis prediction in the form of categorical diffusion, which allows the entire output SMILES sequence to be predicted in unison. We construct an ensemble of diffusion models which achieves state-of-the-art performance for top-1 accuracy and competitive performance for top-3, top-5, and top-10 accuracy among template-free methods. We prove that DiffER is a strong baseline for a new class of template-free model, capable of learning a variety of synthetic techniques used in laboratory settings and outperforming a variety of other template-free methods on top-k accuracy metrics. By constructing an ensemble of categorical diffusion models with a novel length prediction component with variance, our method is able to approximately sample from the posterior distribution of reactants, producing results with strong metrics of confidence and likelihood. Furthermore, our analyses demonstrate that accurate prediction of the SMILES sequence length is key to further boosting the performance of categorical diffusion models.
Analyzing Fine-Grained Alignment and Enhancing Vision Understanding in Multimodal Language Models
May 22, 2025Achieving better alignment between vision embeddings and Large Language Models (LLMs) is crucial for enhancing the abilities of Multimodal LLMs (MLLMs), particularly for recent models that rely on powerful pretrained vision encoders and LLMs. A common approach to connect the pretrained vision encoder and LLM is through a projector applied after the vision encoder. However, the projector is often trained to enable the LLM to generate captions, and hence the mechanism by which LLMs understand each vision token remains unclear. In this work, we first investigate the role of the projector in compressing vision embeddings and aligning them with word embeddings. We show that the projector significantly compresses visual information, removing redundant details while preserving essential elements necessary for the LLM to understand visual content. We then examine patch-level alignment -- the alignment between each vision patch and its corresponding semantic words -- and propose a *multi-semantic alignment hypothesis*. Our analysis indicates that the projector trained by caption loss improves patch-level alignment but only to a limited extent, resulting in weak and coarse alignment. To address this issue, we propose *patch-aligned training* to efficiently enhance patch-level alignment. Our experiments show that patch-aligned training (1) achieves stronger compression capability and improved patch-level alignment, enabling the MLLM to generate higher-quality captions, (2) improves the MLLM's performance by 16% on referring expression grounding tasks, 4% on question-answering tasks, and 3% on modern instruction-following benchmarks when using the same supervised fine-tuning (SFT) setting. The proposed method can be easily extended to other multimodal models.
Planning with Diffusion Models for Target-Oriented Dialogue Systems
Apr 23, 2025Target-Oriented Dialogue (TOD) remains a significant challenge in the LLM era, where strategic dialogue planning is crucial for directing conversations toward specific targets. However, existing dialogue planning methods generate dialogue plans in a step-by-step sequential manner, and may suffer from compounding errors and myopic actions. To address these limitations, we introduce a novel dialogue planning framework, DiffTOD, which leverages diffusion models to enable non-sequential dialogue planning. DiffTOD formulates dialogue planning as a trajectory generation problem with conditional guidance, and leverages a diffusion language model to estimate the likelihood of the dialogue trajectory. To optimize the dialogue action strategies, DiffTOD introduces three tailored guidance mechanisms for different target types, offering flexible guidance towards diverse TOD targets at test time. Extensive experiments across three diverse TOD settings show that DiffTOD can effectively perform non-myopic lookahead exploration and optimize action strategies over a long horizon through non-sequential dialogue planning, and demonstrates strong flexibility across complex and diverse dialogue scenarios. Our code and data are accessible through https://anonymous.4open.science/r/DiffTOD.
LIDDIA: Language-based Intelligent Drug Discovery Agent
Feb 19, 2025



Drug discovery is a long, expensive, and complex process, relying heavily on human medicinal chemists, who can spend years searching the vast space of potential therapies. Recent advances in artificial intelligence for chemistry have sought to expedite individual drug discovery tasks; however, there remains a critical need for an intelligent agent that can navigate the drug discovery process. Towards this end, we introduce LIDDiA, an autonomous agent capable of intelligently navigating the drug discovery process in silico. By leveraging the reasoning capabilities of large language models, LIDDiA serves as a low-cost and highly-adaptable tool for autonomous drug discovery. We comprehensively examine LIDDiA, demonstrating that (1) it can generate molecules meeting key pharmaceutical criteria on over 70% of 30 clinically relevant targets, (2) it intelligently balances exploration and exploitation in the chemical space, and (3) it can identify promising novel drug candidates on EGFR, a critical target for cancers.
$\mathtt{GeLLM^3O}$: Generalizing Large Language Models for Multi-property Molecule Optimization
Feb 19, 2025



Despite recent advancements, most computational methods for molecule optimization are constrained to single- or double-property optimization tasks and suffer from poor scalability and generalizability to novel optimization tasks. Meanwhile, Large Language Models (LLMs) demonstrate remarkable out-of-domain generalizability to novel tasks. To demonstrate LLMs' potential for molecule optimization, we introduce $\mathtt{MoMUInstruct}$, the first high-quality instruction-tuning dataset specifically focused on complex multi-property molecule optimization tasks. Leveraging $\mathtt{MoMUInstruct}$, we develop $\mathtt{GeLLM^3O}$s, a series of instruction-tuned LLMs for molecule optimization. Extensive evaluations across 5 in-domain and 5 out-of-domain tasks demonstrate that $\mathtt{GeLLM^3O}$s consistently outperform state-of-the-art baselines. $\mathtt{GeLLM^3O}$s also exhibit outstanding zero-shot generalization to unseen tasks, significantly outperforming powerful closed-source LLMs. Such strong generalizability demonstrates the tremendous potential of $\mathtt{GeLLM^3O}$s as foundational models for molecule optimization, thereby tackling novel optimization tasks without resource-intensive retraining. $\mathtt{MoMUInstruct}$, models, and code are accessible through https://github.com/ninglab/GeLLMO.
Generating 3D Binding Molecules Using Shape-Conditioned Diffusion Models with Guidance
Feb 09, 2025Drug development is a critical but notoriously resource- and time-consuming process. In this manuscript, we develop a novel generative artificial intelligence (genAI) method DiffSMol to facilitate drug development. DiffSmol generates 3D binding molecules based on the shapes of known ligands. DiffSMol encapsulates geometric details of ligand shapes within pre-trained, expressive shape embeddings and then generates new binding molecules through a diffusion model. DiffSMol further modifies the generated 3D structures iteratively via shape guidance to better resemble the ligand shapes. It also tailors the generated molecules toward optimal binding affinities under the guidance of protein pockets. Here, we show that DiffSMol outperforms the state-of-the-art methods on benchmark datasets. When generating binding molecules resembling ligand shapes, DiffSMol with shape guidance achieves a success rate 61.4%, substantially outperforming the best baseline (11.2%), meanwhile producing molecules with novel molecular graph structures. DiffSMol with pocket guidance also outperforms the best baseline in binding affinities by 13.2%, and even by 17.7% when combined with shape guidance. Case studies for two critical drug targets demonstrate very favorable physicochemical and pharmacokinetic properties of the generated molecules, thus, the potential of DiffSMol in developing promising drug candidates.
Tooling or Not Tooling? The Impact of Tools on Language Agents for Chemistry Problem Solving
Nov 11, 2024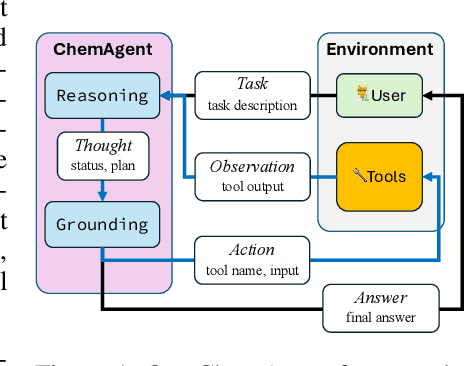
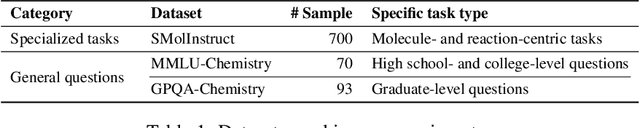

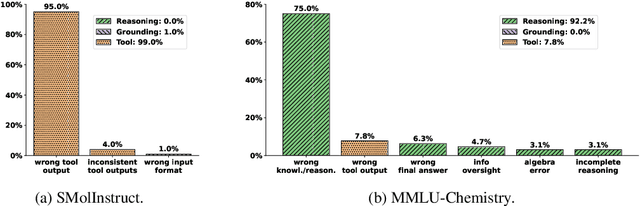
To enhance large language models (LLMs) for chemistry problem solving, several LLM-based agents augmented with tools have been proposed, such as ChemCrow and Coscientist. However, their evaluations are narrow in scope, leaving a large gap in understanding the benefits of tools across diverse chemistry tasks. To bridge this gap, we develop ChemAgent, an enhanced chemistry agent over ChemCrow, and conduct a comprehensive evaluation of its performance on both specialized chemistry tasks and general chemistry questions. Surprisingly, ChemAgent does not consistently outperform its base LLMs without tools. Our error analysis with a chemistry expert suggests that: For specialized chemistry tasks, such as synthesis prediction, we should augment agents with specialized tools; however, for general chemistry questions like those in exams, agents' ability to reason correctly with chemistry knowledge matters more, and tool augmentation does not always help.