 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeArtificial Intelligence for In Silico Clinical Trials: A Review
Sep 16, 2022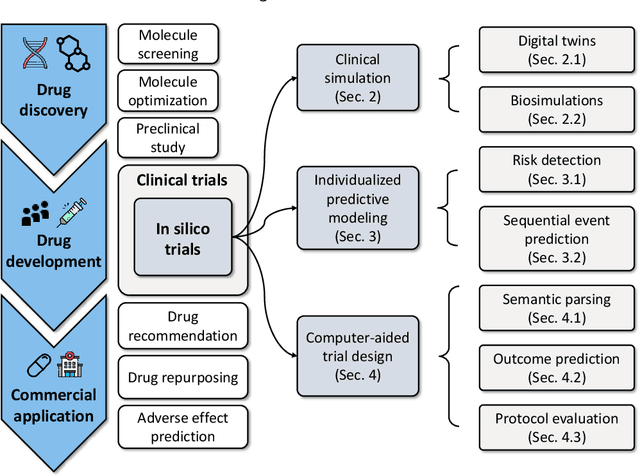
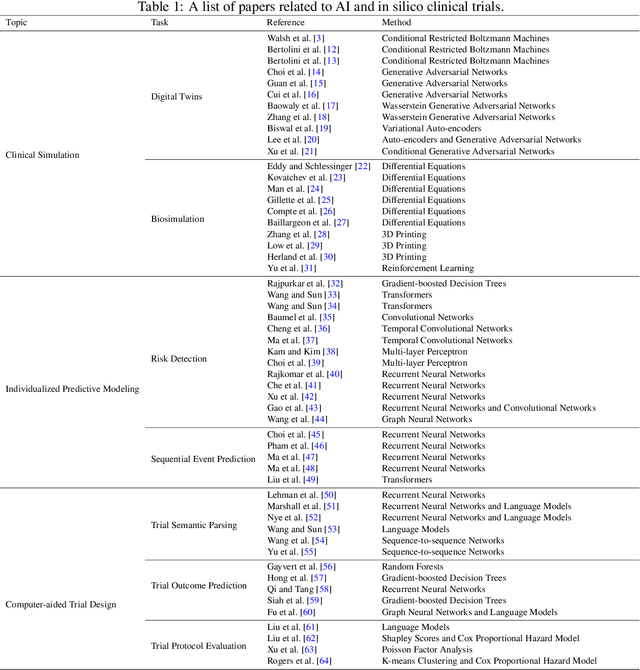
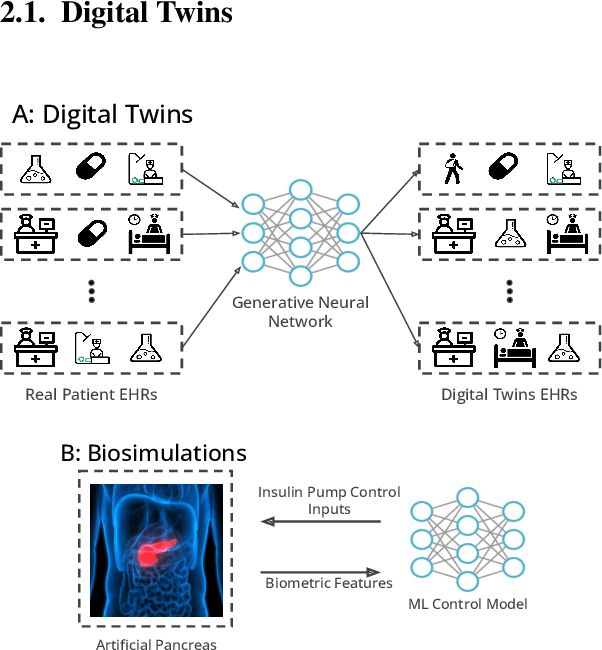
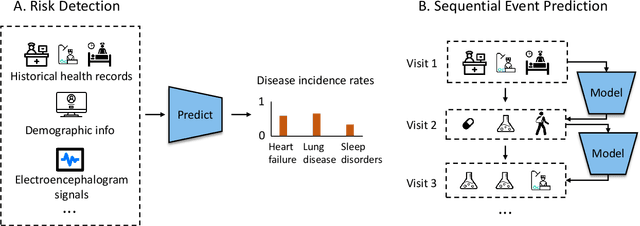
A clinical trial is an essential step in drug development, which is often costly and time-consuming. In silico trials are clinical trials conducted digitally through simulation and modeling as an alternative to traditional clinical trials. AI-enabled in silico trials can increase the case group size by creating virtual cohorts as controls. In addition, it also enables automation and optimization of trial design and predicts the trial success rate. This article systematically reviews papers under three main topics: clinical simulation, individualized predictive modeling, and computer-aided trial design. We focus on how machine learning (ML) may be applied in these applications. In particular, we present the machine learning problem formulation and available data sources for each task. We end with discussing the challenges and opportunities of AI for in silico trials in real-world applications.
Machine Learning Applications for Therapeutic Tasks with Genomics Data
May 03, 2021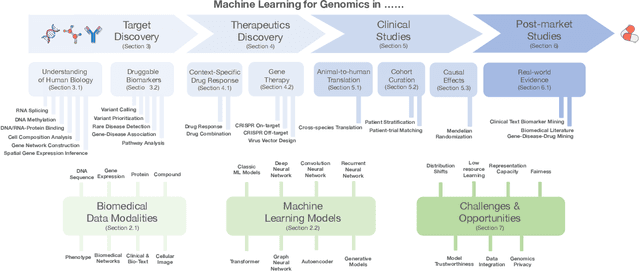
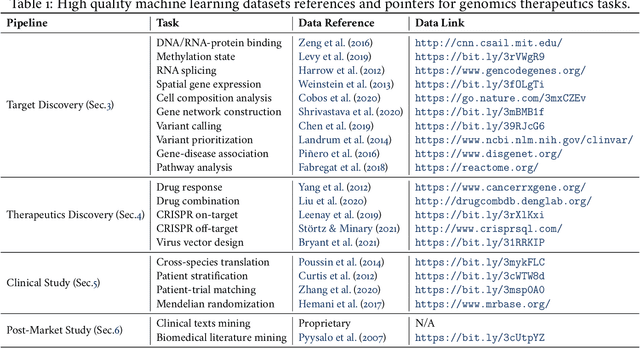
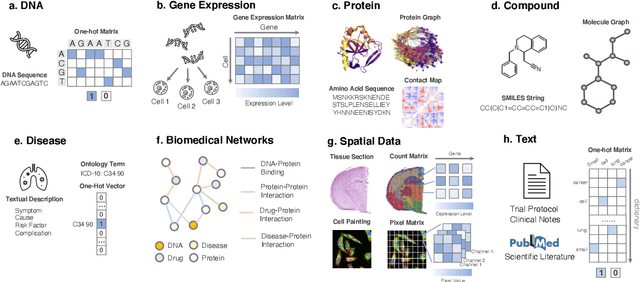
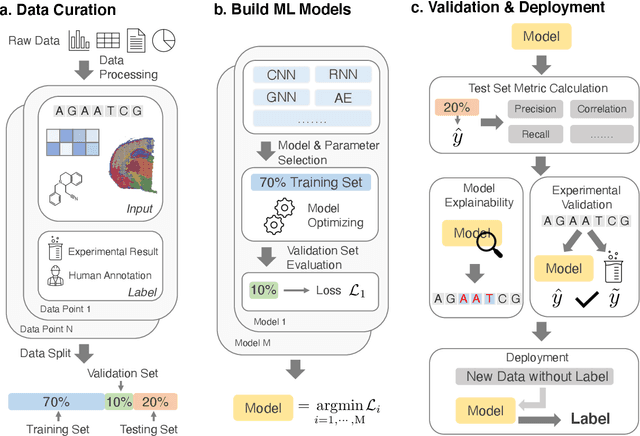
Thanks to the increasing availability of genomics and other biomedical data, many machine learning approaches have been proposed for a wide range of therapeutic discovery and development tasks. In this survey, we review the literature on machine learning applications for genomics through the lens of therapeutic development. We investigate the interplay among genomics, compounds, proteins, electronic health records (EHR), cellular images, and clinical texts. We identify twenty-two machine learning in genomics applications across the entire therapeutics pipeline, from discovering novel targets, personalized medicine, developing gene-editing tools all the way to clinical trials and post-market studies. We also pinpoint seven important challenges in this field with opportunities for expansion and impact. This survey overviews recent research at the intersection of machine learning, genomics, and therapeutic development.
HINT: Hierarchical Interaction Network for Trial Outcome Prediction Leveraging Web Data
Feb 08, 2021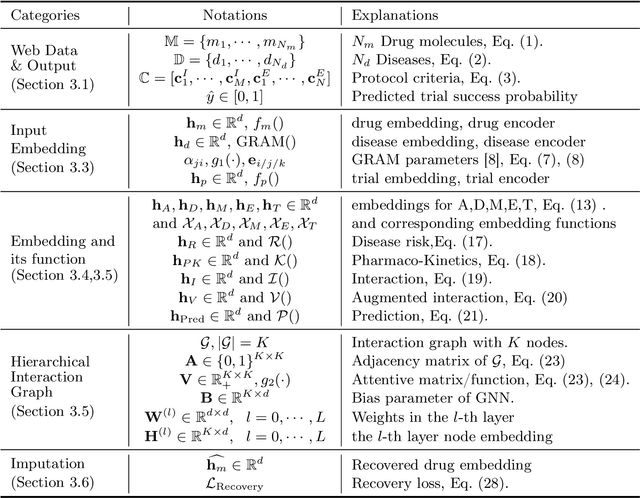
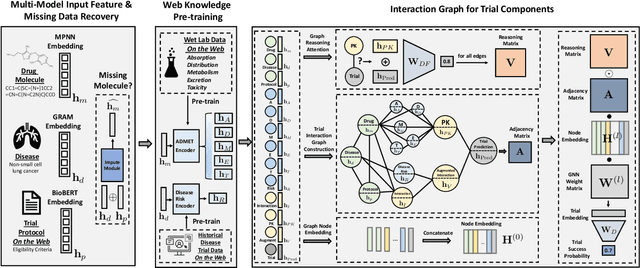
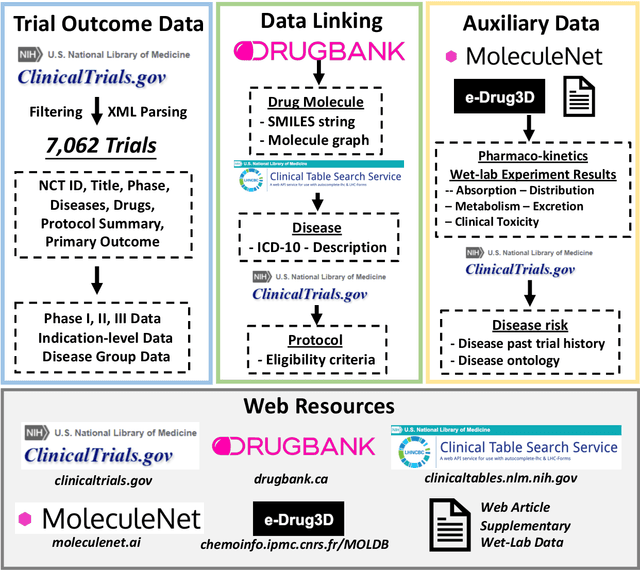
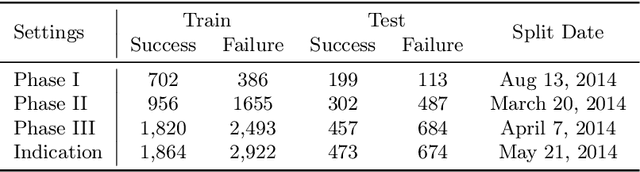
Clinical trials are crucial for drug development but are time consuming, expensive, and often burdensome on patients. More importantly, clinical trials face uncertain outcomes due to issues with efficacy, safety, or problems with patient recruitment. If we were better at predicting the results of clinical trials, we could avoid having to run trials that will inevitably fail more resources could be devoted to trials that are likely to succeed. In this paper, we propose Hierarchical INteraction Network (HINT) for more general, clinical trial outcome predictions for all diseases based on a comprehensive and diverse set of web data including molecule information of the drugs, target disease information, trial protocol and biomedical knowledge. HINT first encode these multi-modal data into latent embeddings, where an imputation module is designed to handle missing data. Next, these embeddings will be fed into the knowledge embedding module to generate knowledge embeddings that are pretrained using external knowledge on pharmaco-kinetic properties and trial risk from the web. Then the interaction graph module will connect all the embedding via domain knowledge to fully capture various trial components and their complex relations as well as their influences on trial outcomes. Finally, HINT learns a dynamic attentive graph neural network to predict trial outcome. Comprehensive experimental results show that HINT achieves strong predictive performance, obtaining 0.772, 0.607, 0.623, 0.703 on PR-AUC for Phase I, II, III, and indication outcome prediction, respectively. It also consistently outperforms the best baseline method by up to 12.4\% on PR-AUC.
STELAR: Spatio-temporal Tensor Factorization with Latent Epidemiological Regularization
Dec 08, 2020
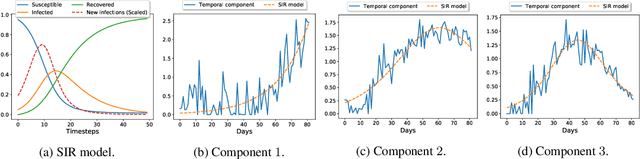
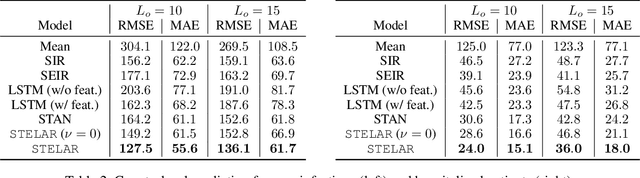
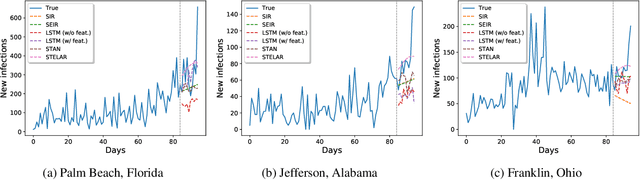
Accurate prediction of the transmission of epidemic diseases such as COVID-19 is crucial for implementing effective mitigation measures. In this work, we develop a tensor method to predict the evolution of epidemic trends for many regions simultaneously. We construct a 3-way spatio-temporal tensor (location, attribute, time) of case counts and propose a nonnegative tensor factorization with latent epidemiological model regularization named STELAR. Unlike standard tensor factorization methods which cannot predict slabs ahead, STELAR enables long-term prediction by incorporating latent temporal regularization through a system of discrete-time difference equations of a widely adopted epidemiological model. We use latent instead of location/attribute-level epidemiological dynamics to capture common epidemic profile sub-types and improve collaborative learning and prediction. We conduct experiments using both county- and state-level COVID-19 data and show that our model can identify interesting latent patterns of the epidemic. Finally, we evaluate the predictive ability of our method and show superior performance compared to the baselines, achieving up to 21% lower root mean square error and 25% lower mean absolute error for county-level prediction.
FLANNEL: Focal Loss Based Neural Network Ensemble for COVID-19 Detection
Oct 30, 2020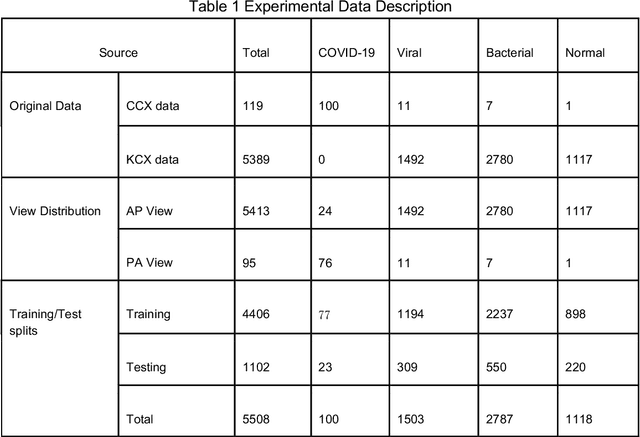
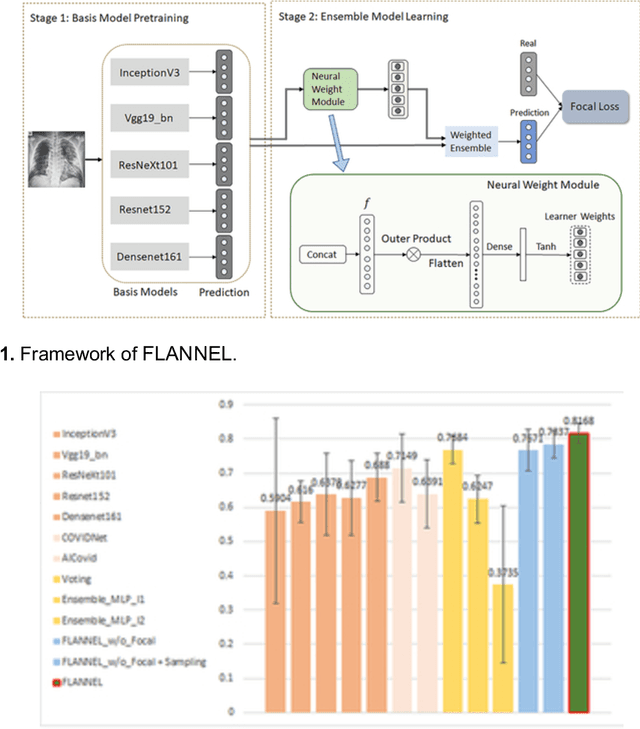
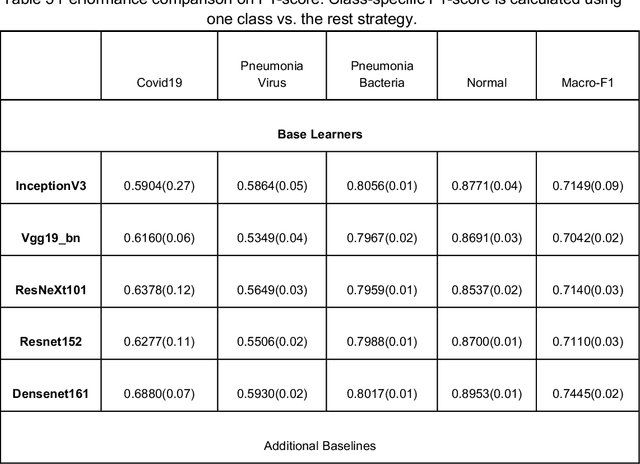
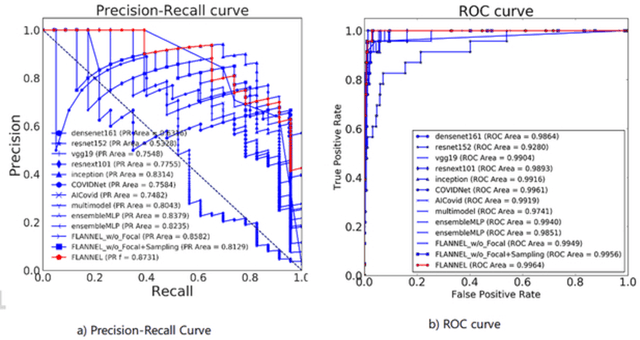
To test the possibility of differentiating chest x-ray images of COVID-19 against other pneumonia and healthy patients using deep neural networks. We construct the X-ray imaging data from two publicly available sources, which include 5508 chest x-ray images across 2874 patients with four classes: normal, bacterial pneumonia, non-COVID-19 viral pneumonia, and COVID-19. To identify COVID-19, we propose a Focal Loss Based Neural Ensemble Network (FLANNEL), a flexible module to ensemble several convolutional neural network (CNN) models and fuse with a focal loss for accurate COVID-19 detection on class imbalance data. FLANNEL consistently outperforms baseline models on COVID-19 identification task in all metrics. Compared with the best baseline, FLANNEL shows a higher macro-F1 score with 6% relative increase on Covid-19 identification task where it achieves 0.7833(0.07) in Precision, 0.8609(0.03) in Recall, and 0.8168(0.03) F1 score.
UNITE: Uncertainty-based Health Risk Prediction Leveraging Multi-sourced Data
Oct 22, 2020
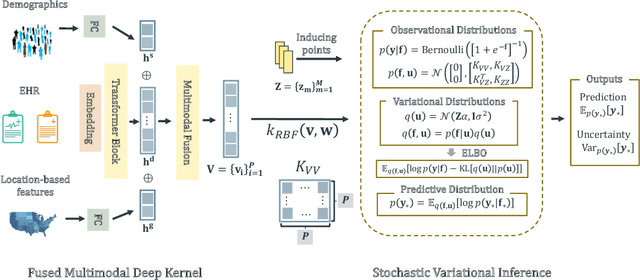
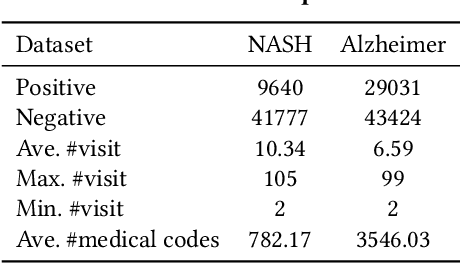
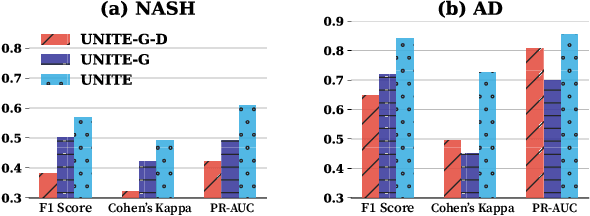
Successful health risk prediction demands accuracy and reliability of the model. Existing predictive models mainly depend on mining electronic health records (EHR) with advanced deep learning techniques to improve model accuracy. However, they all ignore the importance of publicly available online health data, especially socioeconomic status, environmental factors, and detailed demographic information for each location, which are all strong predictive signals and can definitely augment precision medicine. To achieve model reliability, the model needs to provide accurate prediction and uncertainty score of the prediction. However, existing uncertainty estimation approaches often failed in handling high-dimensional data, which are present in multi-sourced data. To fill the gap, we propose UNcertaInTy-based hEalth risk prediction (UNITE) model. Building upon an adaptive multimodal deep kernel and a stochastic variational inference module, UNITE provides accurate disease risk prediction and uncertainty estimation leveraging multi-sourced health data including EHR data, patient demographics, and public health data collected from the web. We evaluate UNITE on real-world disease risk prediction tasks: nonalcoholic fatty liver disease (NASH) and Alzheimer's disease (AD). UNITE achieves up to 0.841 in F1 score for AD detection, up to 0.609 in PR-AUC for NASH detection, and outperforms various state-of-the-art baselines by up to $19\%$ over the best baseline. We also show UNITE can model meaningful uncertainties and can provide evidence-based clinical support by clustering similar patients.
MolDesigner: Interactive Design of Efficacious Drugs with Deep Learning
Oct 05, 2020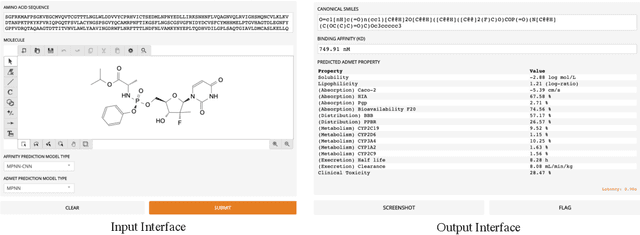
The efficacy of a drug depends on its binding affinity to the therapeutic target and pharmacokinetics. Deep learning (DL) has demonstrated remarkable progress in predicting drug efficacy. We develop MolDesigner, a human-in-the-loop web user-interface (UI), to assist drug developers leverage DL predictions to design more effective drugs. A developer can draw a drug molecule in the interface. In the backend, more than 17 state-of-the-art DL models generate predictions on important indices that are crucial for a drug's efficacy. Based on these predictions, drug developers can edit the drug molecule and reiterate until satisfaction. MolDesigner can make predictions in real-time with a latency of less than a second.
MIMOSA: Multi-constraint Molecule Sampling for Molecule Optimization
Oct 05, 2020
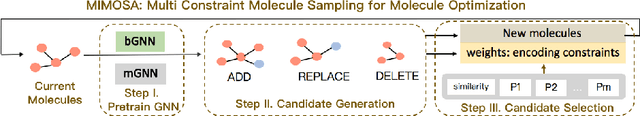
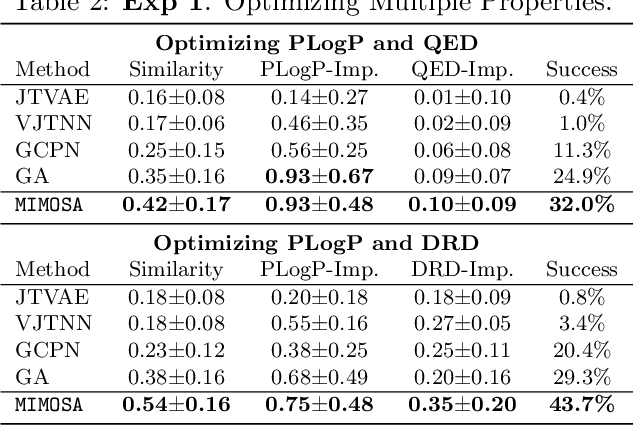
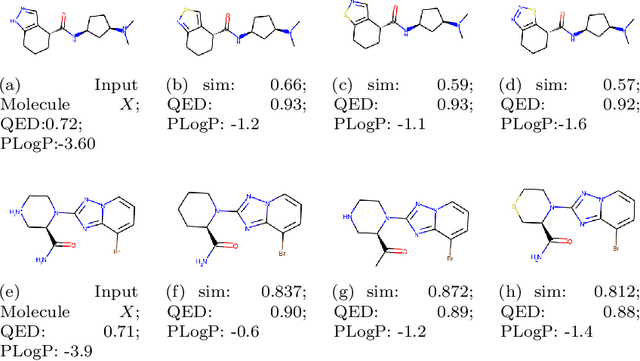
Molecule optimization is a fundamental task for accelerating drug discovery, with the goal of generating new valid molecules that maximize multiple drug properties while maintaining similarity to the input molecule. Existing generative models and reinforcement learning approaches made initial success, but still face difficulties in simultaneously optimizing multiple drug properties. To address such challenges, we propose the MultI-constraint MOlecule SAmpling (MIMOSA) approach, a sampling framework to use input molecule as an initial guess and sample molecules from the target distribution. MIMOSA first pretrains two property agnostic graph neural networks (GNNs) for molecule topology and substructure-type prediction, where a substructure can be either atom or single ring. For each iteration, MIMOSA uses the GNNs' prediction and employs three basic substructure operations (add, replace, delete) to generate new molecules and associated weights. The weights can encode multiple constraints including similarity and drug property constraints, upon which we select promising molecules for next iteration. MIMOSA enables flexible encoding of multiple property- and similarity-constraints and can efficiently generate new molecules that satisfy various property constraints and achieved up to 49.6% relative improvement over the best baseline in terms of success rate.
SumGNN: Multi-typed Drug Interaction Prediction via Efficient Knowledge Graph Summarization
Oct 04, 2020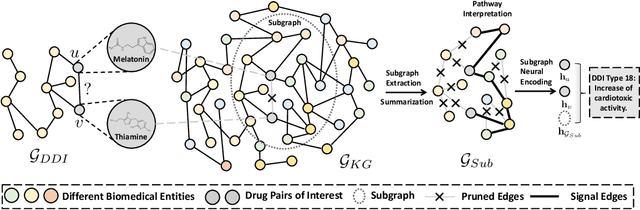
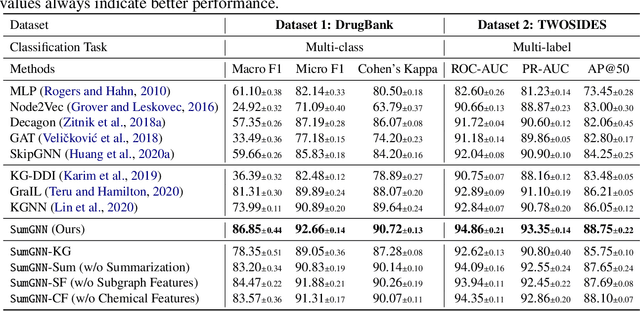
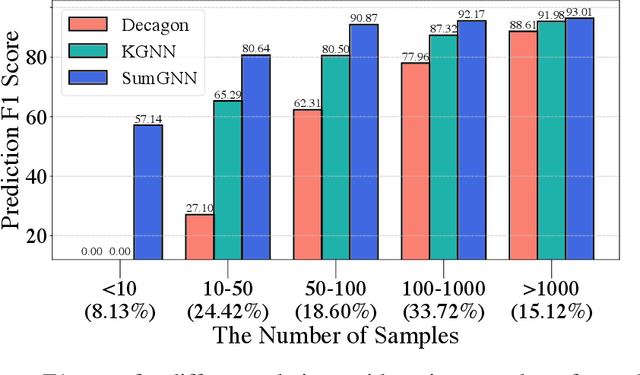
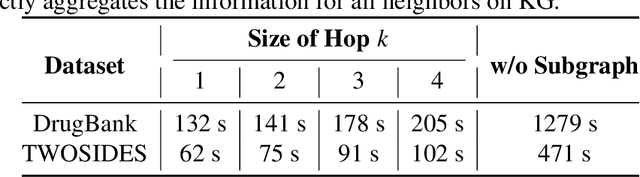
Thanks to the increasing availability of drug-drug interactions (DDI) datasets and large biomedical knowledge graphs (KGs), accurate detection of adverse DDI using machine learning models becomes possible. However, it remains largely an open problem how to effectively utilize large and noisy biomedical KG for DDI detection. Due to its sheer size and amount of noise in KGs, it is often less beneficial to directly integrate KGs with other smaller but higher quality data (e.g., experimental data). Most of existing approaches ignore KGs altogether. Some tries to directly integrate KGs with other data via graph neural networks with limited success. Furthermore most previous works focus on binary DDI prediction whereas the multi-typed DDI pharmacological effect prediction is more meaningful but harder task. To fill the gaps, we propose a new method SumGNN:~{\it knowledge summarization graph neural network}, which is enabled by a subgraph extraction module that can efficiently anchor on relevant subgraphs from a KG, a self-attention based subgraph summarization scheme to generate reasoning path within the subgraph, and a multi-channel knowledge and data integration module that utilizes massive external biomedical knowledge for significantly improved multi-typed DDI predictions. SumGNN outperforms the best baseline by up to 5.54\%, and performance gain is particularly significant in low data relation types. In addition, SumGNN provides interpretable prediction via the generated reasoning paths for each prediction.
COMPOSE: Cross-Modal Pseudo-Siamese Network for Patient Trial Matching
Jun 15, 2020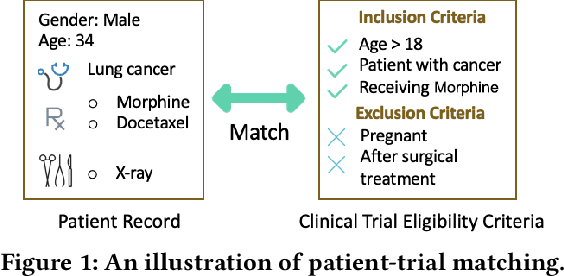
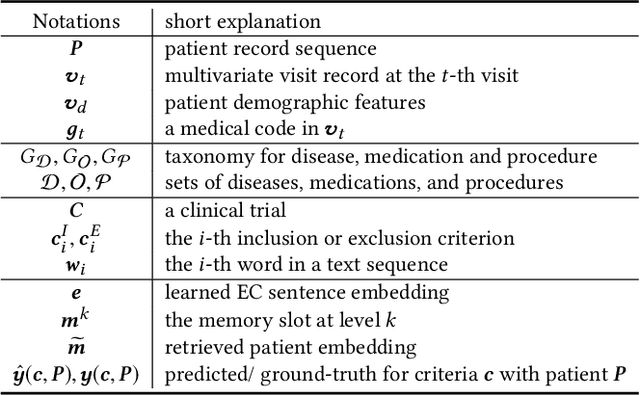
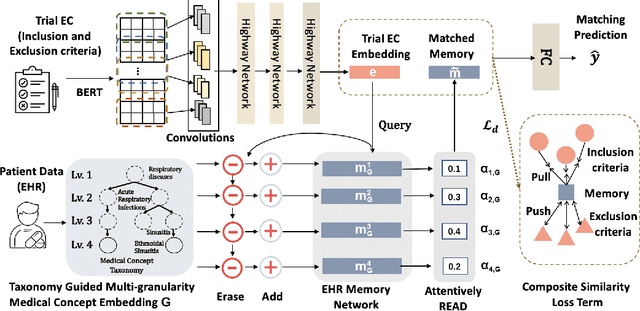
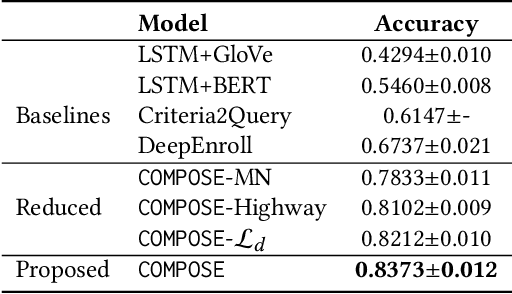
Clinical trials play important roles in drug development but often suffer from expensive, inaccurate and insufficient patient recruitment. The availability of massive electronic health records (EHR) data and trial eligibility criteria (EC) bring a new opportunity to data driven patient recruitment. One key task named patient-trial matching is to find qualified patients for clinical trials given structured EHR and unstructured EC text (both inclusion and exclusion criteria). How to match complex EC text with longitudinal patient EHRs? How to embed many-to-many relationships between patients and trials? How to explicitly handle the difference between inclusion and exclusion criteria? In this paper, we proposed CrOss-Modal PseudO-SiamEse network (COMPOSE) to address these challenges for patient-trial matching. One path of the network encodes EC using convolutional highway network. The other path processes EHR with multi-granularity memory network that encodes structured patient records into multiple levels based on medical ontology. Using the EC embedding as query, COMPOSE performs attentional record alignment and thus enables dynamic patient-trial matching. COMPOSE also introduces a composite loss term to maximize the similarity between patient records and inclusion criteria while minimize the similarity to the exclusion criteria. Experiment results show COMPOSE can reach 98.0% AUC on patient-criteria matching and 83.7% accuracy on patient-trial matching, which leads 24.3% improvement over the best baseline on real-world patient-trial matching tasks.