 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMIMOSA: Multi-constraint Molecule Sampling for Molecule Optimization
Paper and Code

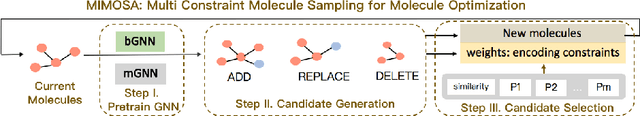
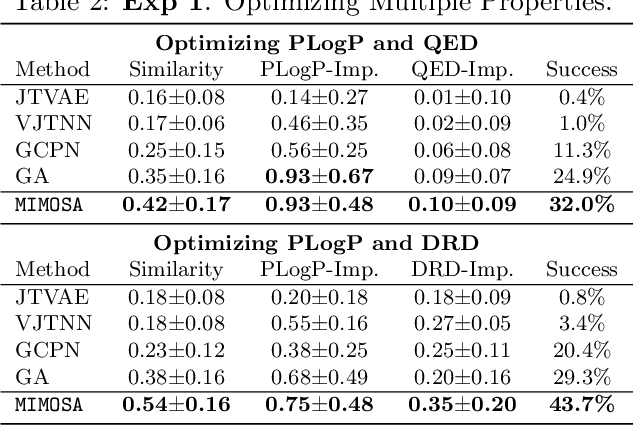
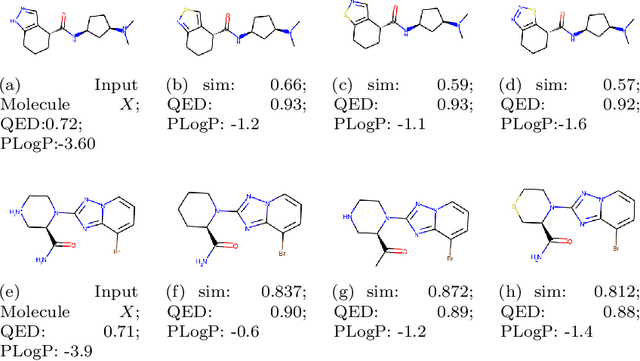
Molecule optimization is a fundamental task for accelerating drug discovery, with the goal of generating new valid molecules that maximize multiple drug properties while maintaining similarity to the input molecule. Existing generative models and reinforcement learning approaches made initial success, but still face difficulties in simultaneously optimizing multiple drug properties. To address such challenges, we propose the MultI-constraint MOlecule SAmpling (MIMOSA) approach, a sampling framework to use input molecule as an initial guess and sample molecules from the target distribution. MIMOSA first pretrains two property agnostic graph neural networks (GNNs) for molecule topology and substructure-type prediction, where a substructure can be either atom or single ring. For each iteration, MIMOSA uses the GNNs' prediction and employs three basic substructure operations (add, replace, delete) to generate new molecules and associated weights. The weights can encode multiple constraints including similarity and drug property constraints, upon which we select promising molecules for next iteration. MIMOSA enables flexible encoding of multiple property- and similarity-constraints and can efficiently generate new molecules that satisfy various property constraints and achieved up to 49.6% relative improvement over the best baseline in terms of success rate.