 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCurating a COVID-19 data repository and forecasting county-level death counts in the United States
May 16, 2020
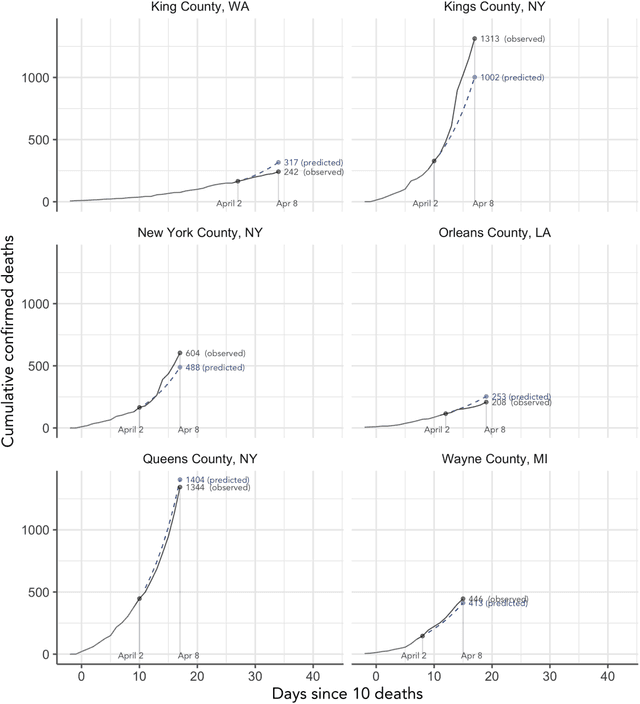
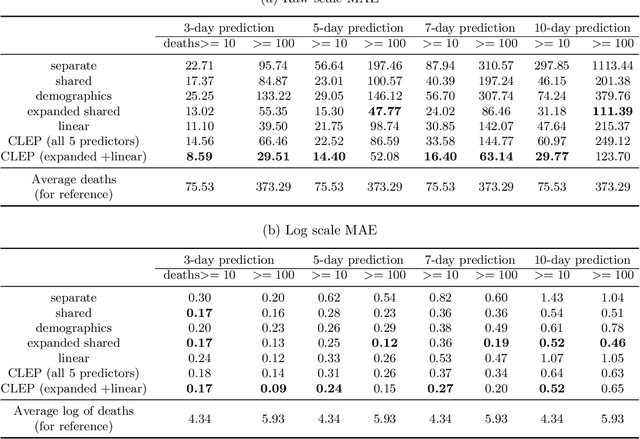
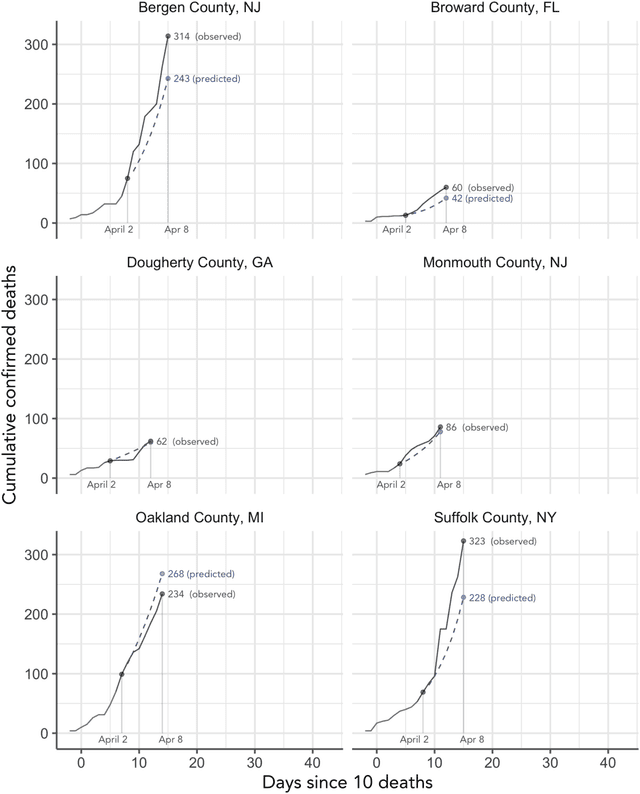
As the COVID-19 outbreak continues to evolve, accurate forecasting continues to play an extremely important role in informing policy decisions. In this paper, we collate a large data repository containing COVID-19 information from a range of different sources. We use this data to develop several predictors and prediction intervals for forecasting the short-term (e.g., over the next week) trajectory of COVID-19-related recorded deaths at the county-level in the United States. Specifically, using data from January 22, 2020, to May 10, 2020, we produce several different predictors and combine their forecasts using ensembling techniques, resulting in an ensemble we refer to as Combined Linear and Exponential Predictors (CLEP). Our individual predictors include county-specific exponential and linear predictors, an exponential predictor that pools data together across counties, and a demographics-based exponential predictor. In addition, we use the largest prediction errors in the past five days to assess the uncertainty of our death predictions, resulting in prediction intervals that we refer to as Maximum (absolute) Error Prediction Intervals (MEPI). We show that MEPI is an effective method in practice with a 94.5\% coverage rate when averaged across counties. Our forecasts are already being used by the non-profit organization, Response4Life, to determine the medical supply need for individual hospitals and have directly contributed to the distribution of medical supplies across the country. We hope that our forecasts and data repository can help guide necessary county-specific decision-making and help counties prepare for their continued fight against COVID-19. All collected data, modeling code, forecasts, and visualizations are updated daily and available at \url{https://github.com/Yu-Group/covid19-severity-prediction}.
A Debiased MDI Feature Importance Measure for Random Forests
Jun 26, 2019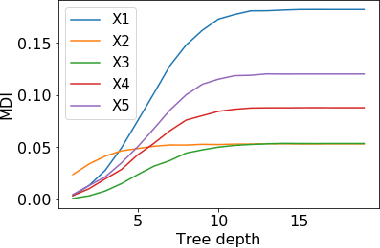
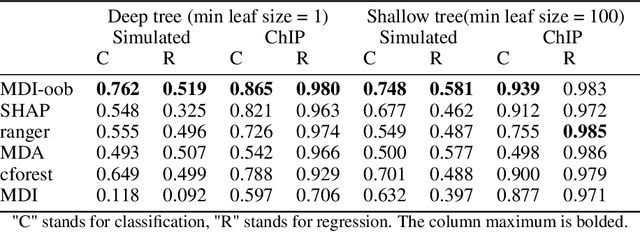
Tree ensembles such as Random Forests have achieved impressive empirical success across a wide variety of applications. To understand how these models make predictions, people routinely turn to feature importance measures calculated from tree ensembles. It has long been known that Mean Decrease Impurity (MDI), one of the most widely used measures of feature importance, incorrectly assigns high importance to noisy features, leading to systematic bias in feature selection. In this paper, we address the feature selection bias of MDI from both theoretical and methodological perspectives. Based on the original definition of MDI by Breiman et al. for a single tree, we derive a tight non-asymptotic bound on the expected bias of MDI importance of noisy features, showing that deep trees have higher (expected) feature selection bias than shallow ones. However, it is not clear how to reduce the bias of MDI using its existing analytical expression. We derive a new analytical expression for MDI, and based on this new expression, we are able to propose a debiased MDI feature importance measure using out-of-bag samples, called MDI-oob. For both the simulated data and a genomic ChIP dataset, MDI-oob achieves state-of-the-art performance in feature selection from Random Forests for both deep and shallow trees.
Three principles of data science: predictability, computability, and stability (PCS)
Jan 23, 2019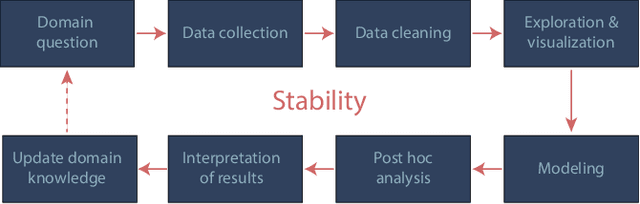
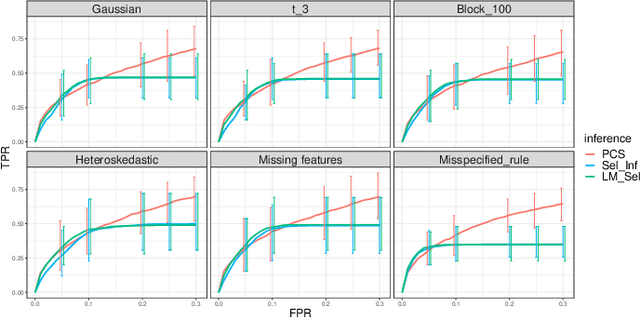
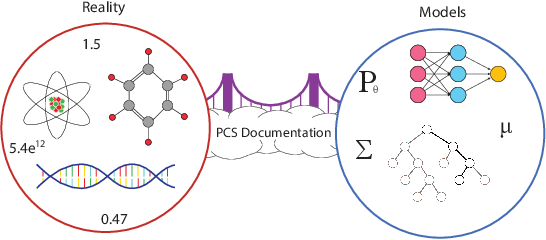
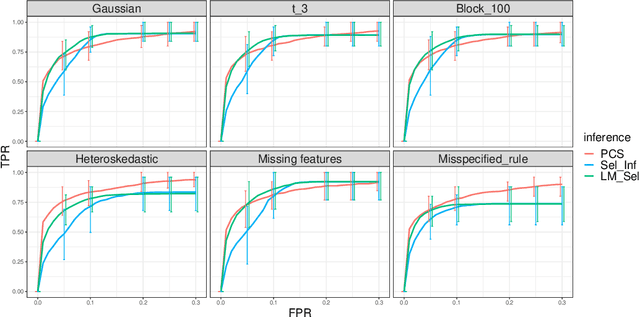
We propose the predictability, computability, and stability (PCS) framework to extract reproducible knowledge from data that can guide scientific hypothesis generation and experimental design. The PCS framework builds on key ideas in machine learning, using predictability as a reality check and evaluating computational considerations in data collection, data storage, and algorithm design. It augments PC with an overarching stability principle, which largely expands traditional statistical uncertainty considerations. In particular, stability assesses how results vary with respect to choices (or perturbations) made across the data science life cycle, including problem formulation, pre-processing, modeling (data and algorithm perturbations), and exploratory data analysis (EDA) before and after modeling. Furthermore, we develop PCS inference to investigate the stability of data results and identify when models are consistent with relatively simple phenomena. We compare PCS inference with existing methods, such as selective inference, in high-dimensional sparse linear model simulations to demonstrate that our methods consistently outperform others in terms of ROC curves over a wide range of simulation settings. Finally, we propose a PCS documentation based on Rmarkdown, iPython, or Jupyter Notebook, with publicly available, reproducible codes and narratives to back up human choices made throughout an analysis. The PCS workflow and documentation are demonstrated in a genomics case study available on Zenodo.
Interpretable machine learning: definitions, methods, and applications
Jan 14, 2019
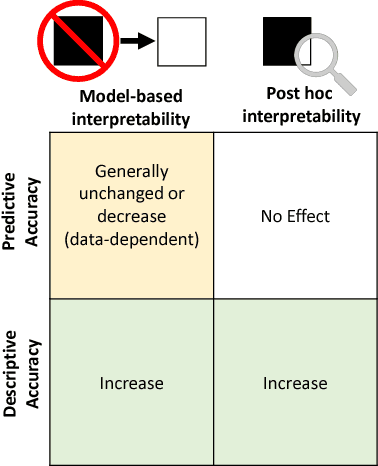
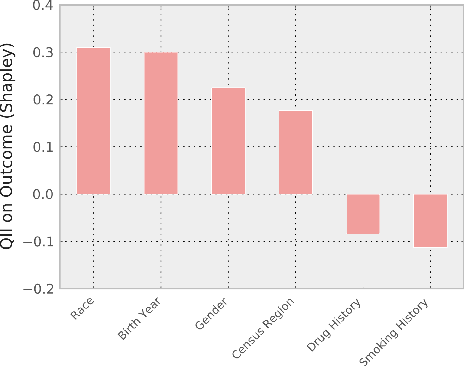
Machine-learning models have demonstrated great success in learning complex patterns that enable them to make predictions about unobserved data. In addition to using models for prediction, the ability to interpret what a model has learned is receiving an increasing amount of attention. However, this increased focus has led to considerable confusion about the notion of interpretability. In particular, it is unclear how the wide array of proposed interpretation methods are related, and what common concepts can be used to evaluate them. We aim to address these concerns by defining interpretability in the context of machine learning and introducing the Predictive, Descriptive, Relevant (PDR) framework for discussing interpretations. The PDR framework provides three overarching desiderata for evaluation: predictive accuracy, descriptive accuracy and relevancy, with relevancy judged relative to a human audience. Moreover, to help manage the deluge of interpretation methods, we introduce a categorization of existing techniques into model-based and post-hoc categories, with sub-groups including sparsity, modularity and simulatability. To demonstrate how practitioners can use the PDR framework to evaluate and understand interpretations, we provide numerous real-world examples. These examples highlight the often under-appreciated role played by human audiences in discussions of interpretability. Finally, based on our framework, we discuss limitations of existing methods and directions for future work. We hope that this work will provide a common vocabulary that will make it easier for both practitioners and researchers to discuss and choose from the full range of interpretation methods.
Refining interaction search through signed iterative Random Forests
Oct 16, 2018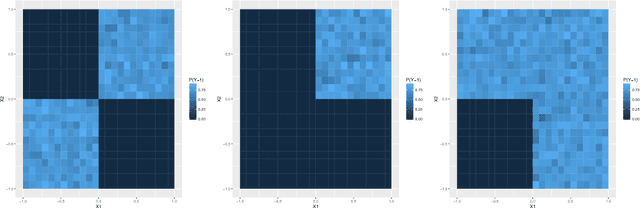
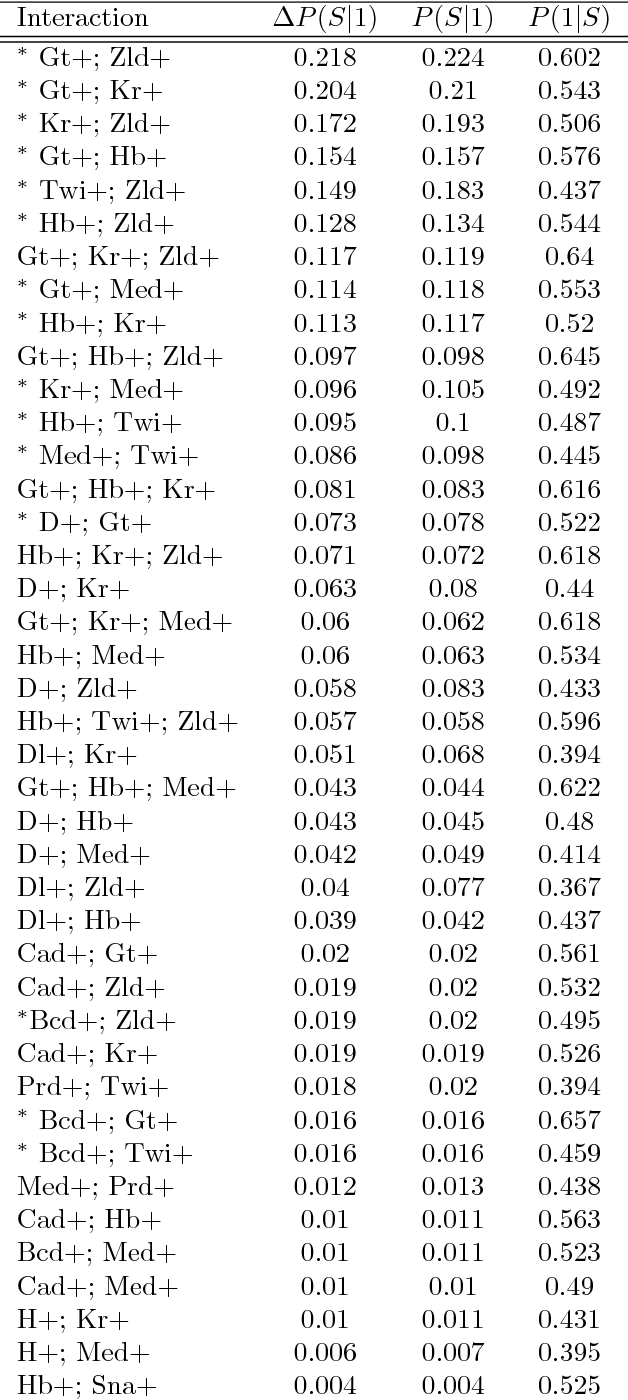
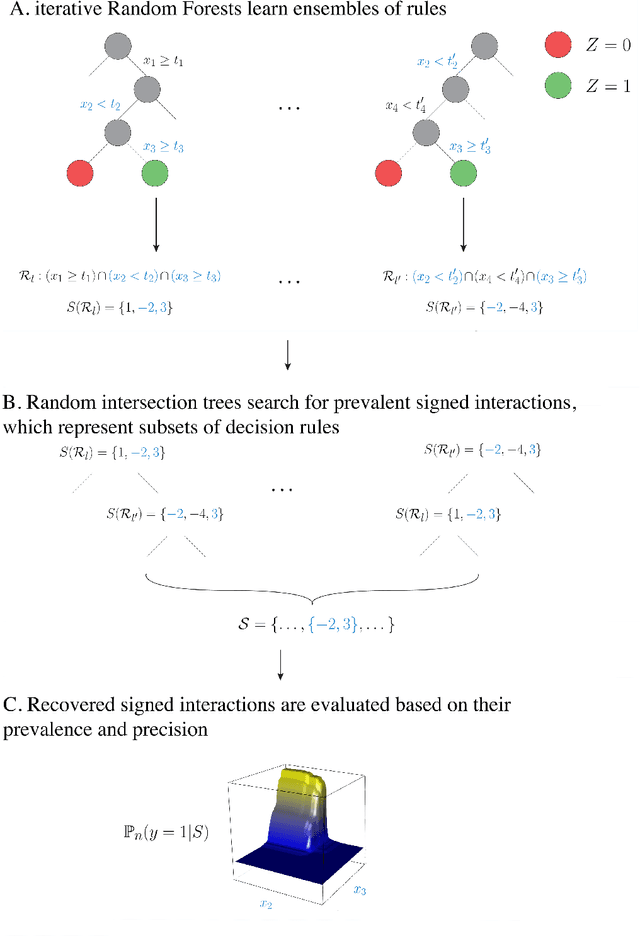
Advances in supervised learning have enabled accurate prediction in biological systems governed by complex interactions among biomolecules. However, state-of-the-art predictive algorithms are typically black-boxes, learning statistical interactions that are difficult to translate into testable hypotheses. The iterative Random Forest algorithm took a step towards bridging this gap by providing a computationally tractable procedure to identify the stable, high-order feature interactions that drive the predictive accuracy of Random Forests (RF). Here we refine the interactions identified by iRF to explicitly map responses as a function of interacting features. Our method, signed iRF, describes subsets of rules that frequently occur on RF decision paths. We refer to these rule subsets as signed interactions. Signed interactions share not only the same set of interacting features but also exhibit similar thresholding behavior, and thus describe a consistent functional relationship between interacting features and responses. We describe stable and predictive importance metrics to rank signed interactions. For each SPIM, we define null importance metrics that characterize its expected behavior under known structure. We evaluate our proposed approach in biologically inspired simulations and two case studies: predicting enhancer activity and spatial gene expression patterns. In the case of enhancer activity, s-iRF recovers one of the few experimentally validated high-order interactions and suggests novel enhancer elements where this interaction may be active. In the case of spatial gene expression patterns, s-iRF recovers all 11 reported links in the gap gene network. By refining the process of interaction recovery, our approach has the potential to guide mechanistic inquiry into systems whose scale and complexity is beyond human comprehension.
Iterative Random Forests to detect predictive and stable high-order interactions
Dec 23, 2017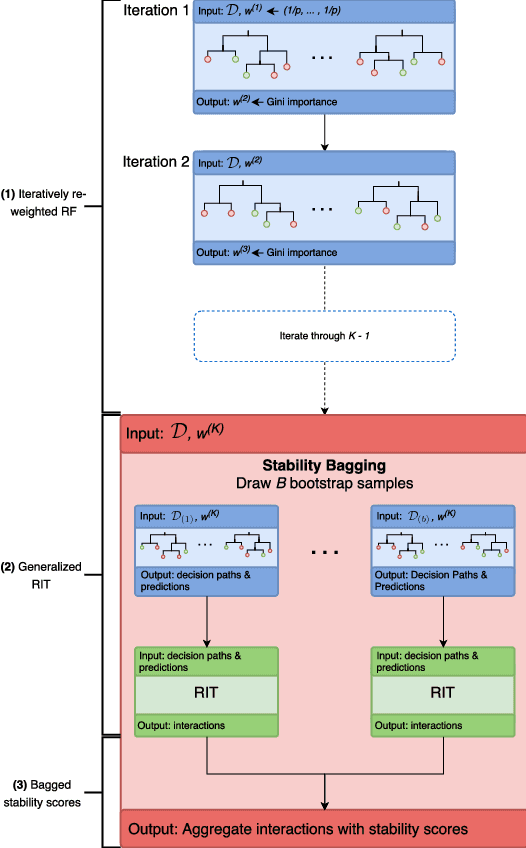
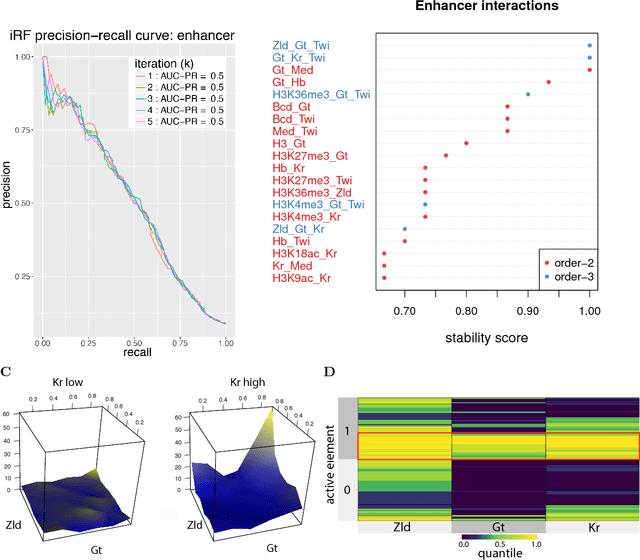
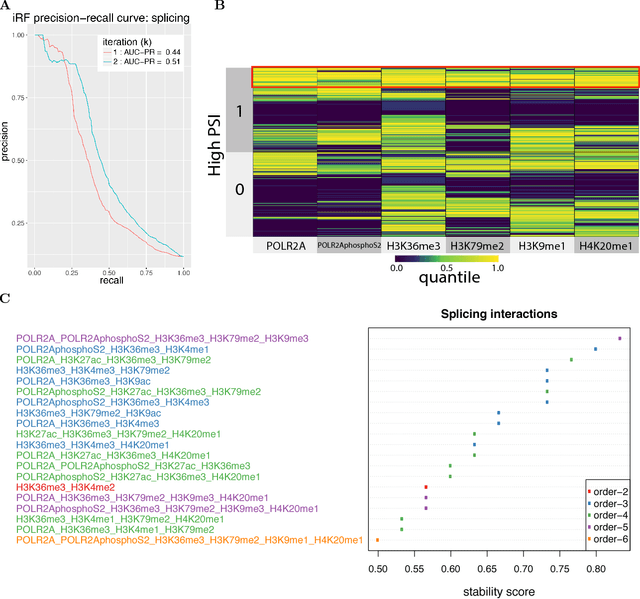
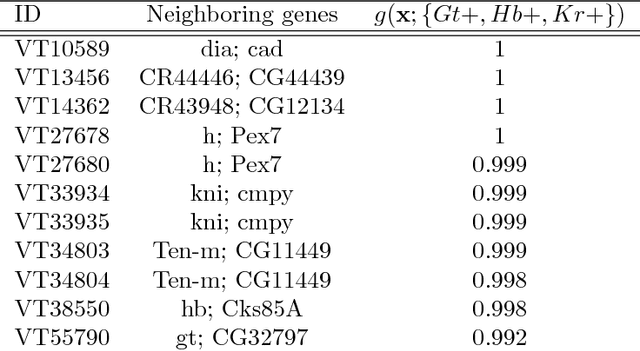
Genomics has revolutionized biology, enabling the interrogation of whole transcriptomes, genome-wide binding sites for proteins, and many other molecular processes. However, individual genomic assays measure elements that interact in vivo as components of larger molecular machines. Understanding how these high-order interactions drive gene expression presents a substantial statistical challenge. Building on Random Forests (RF), Random Intersection Trees (RITs), and through extensive, biologically inspired simulations, we developed the iterative Random Forest algorithm (iRF). iRF trains a feature-weighted ensemble of decision trees to detect stable, high-order interactions with same order of computational cost as RF. We demonstrate the utility of iRF for high-order interaction discovery in two prediction problems: enhancer activity in the early Drosophila embryo and alternative splicing of primary transcripts in human derived cell lines. In Drosophila, among the 20 pairwise transcription factor interactions iRF identifies as stable (returned in more than half of bootstrap replicates), 80% have been previously reported as physical interactions. Moreover, novel third-order interactions, e.g. between Zelda (Zld), Giant (Gt), and Twist (Twi), suggest high-order relationships that are candidates for follow-up experiments. In human-derived cells, iRF re-discovered a central role of H3K36me3 in chromatin-mediated splicing regulation, and identified novel 5th and 6th order interactions, indicative of multi-valent nucleosomes with specific roles in splicing regulation. By decoupling the order of interactions from the computational cost of identification, iRF opens new avenues of inquiry into the molecular mechanisms underlying genome biology.
Artificial Intelligence and Statistics
Dec 08, 2017Artificial intelligence (AI) is intrinsically data-driven. It calls for the application of statistical concepts through human-machine collaboration during generation of data, development of algorithms, and evaluation of results. This paper discusses how such human-machine collaboration can be approached through the statistical concepts of population, question of interest, representativeness of training data, and scrutiny of results (PQRS). The PQRS workflow provides a conceptual framework for integrating statistical ideas with human input into AI products and research. These ideas include experimental design principles of randomization and local control as well as the principle of stability to gain reproducibility and interpretability of algorithms and data results. We discuss the use of these principles in the contexts of self-driving cars, automated medical diagnoses, and examples from the authors' collaborative research.