 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearning from learning machines: a new generation of AI technology to meet the needs of science
Nov 27, 2021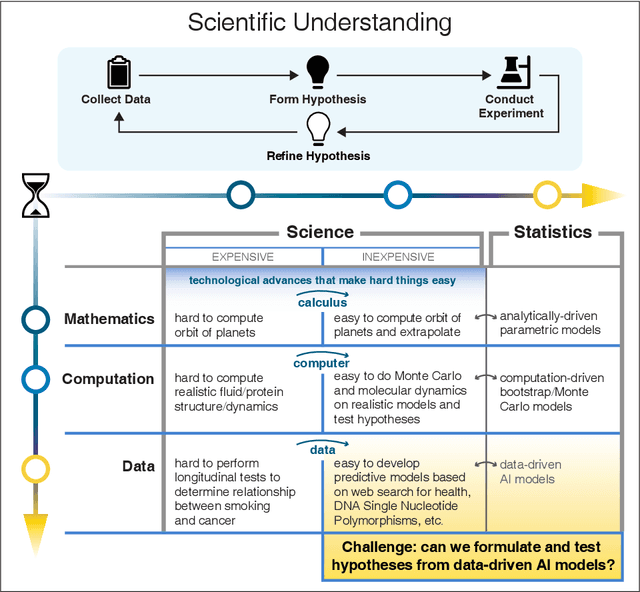

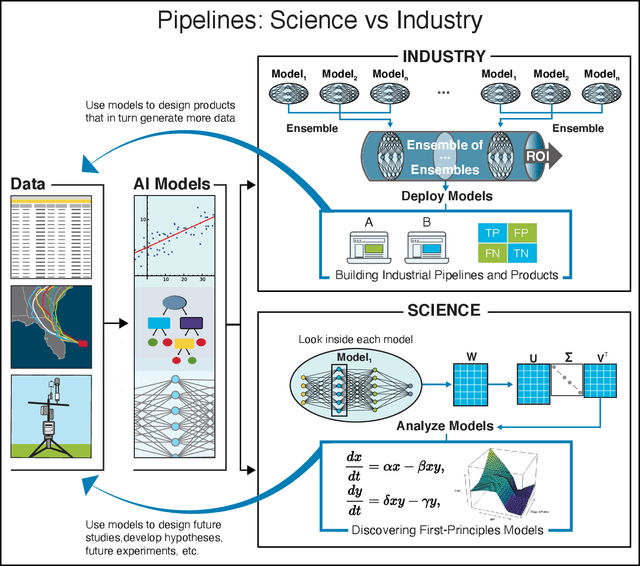
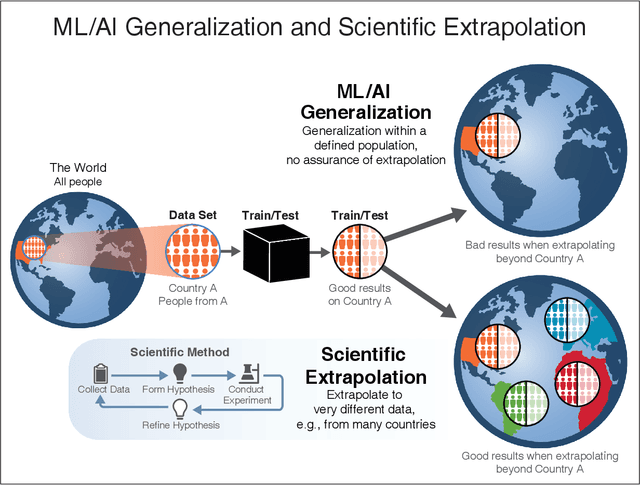
We outline emerging opportunities and challenges to enhance the utility of AI for scientific discovery. The distinct goals of AI for industry versus the goals of AI for science create tension between identifying patterns in data versus discovering patterns in the world from data. If we address the fundamental challenges associated with "bridging the gap" between domain-driven scientific models and data-driven AI learning machines, then we expect that these AI models can transform hypothesis generation, scientific discovery, and the scientific process itself.
Spatial Graph Attention and Curiosity-driven Policy for Antiviral Drug Discovery
Jun 20, 2021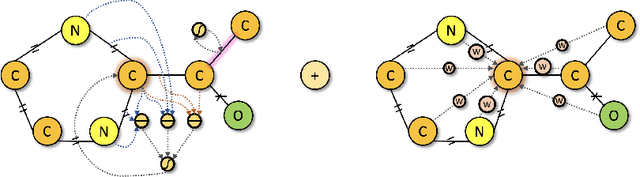
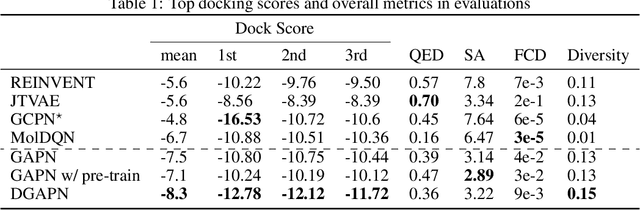
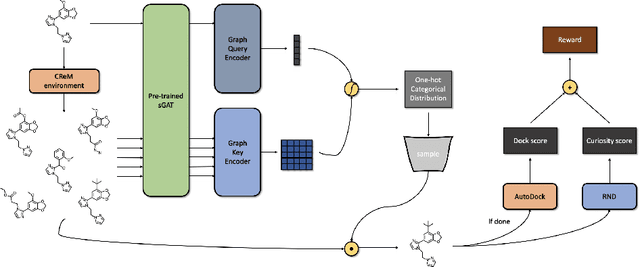
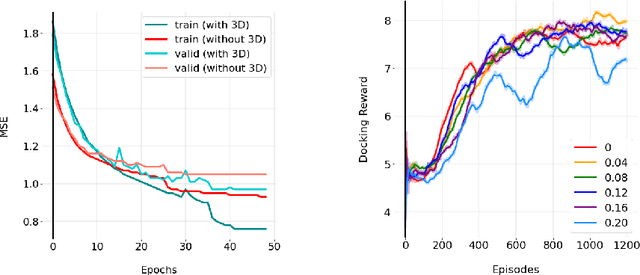
We developed Distilled Graph Attention Policy Networks (DGAPNs), a curiosity-driven reinforcement learning model to generate novel graph-structured chemical representations that optimize user-defined objectives by efficiently navigating a physically constrained domain. The framework is examined on the task of generating molecules that are designed to bind, noncovalently, to functional sites of SARS-CoV-2 proteins. We present a spatial Graph Attention Network (sGAT) that leverages self-attention over both node and edge attributes as well as encoding spatial structure -- this capability is of considerable interest in areas such as molecular and synthetic biology and drug discovery. An attentional policy network is then introduced to learn decision rules for a dynamic, fragment-based chemical environment, and state-of-the-art policy gradient techniques are employed to train the network with enhanced stability. Exploration is efficiently encouraged by incorporating innovation reward bonuses learned and proposed by random network distillation. In experiments, our framework achieved outstanding results compared to state-of-the-art algorithms, while increasing the diversity of proposed molecules and reducing the complexity of paths to chemical synthesis.
Sparse Canonical Correlation Analysis via Concave Minimization
Sep 17, 2019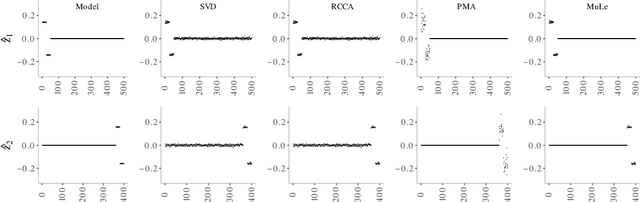
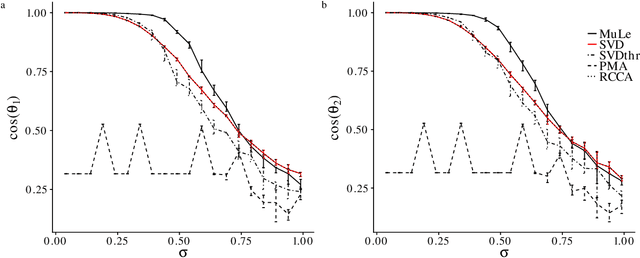
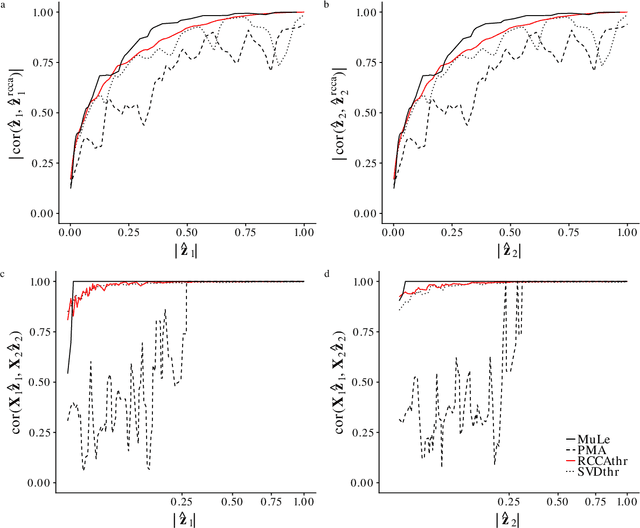
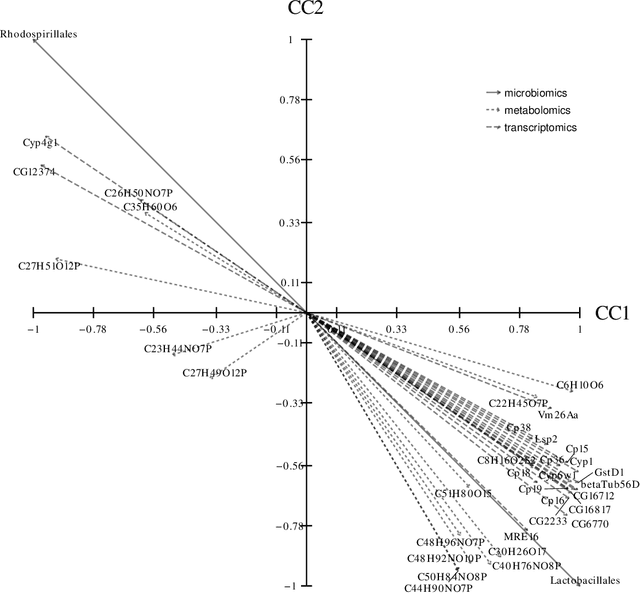
A new approach to the sparse Canonical Correlation Analysis (sCCA)is proposed with the aim of discovering interpretable associations in very high-dimensional multi-view, i.e.observations of multiple sets of variables on the same subjects, problems. Inspired by the sparse PCA approach of Journee et al. (2010), we also show that the sparse CCA formulation, while non-convex, is equivalent to a maximization program of a convex objective over a compact set for which we propose a first-order gradient method. This result helps us reduce the search space drastically to the boundaries of the set. Consequently, we propose a two-step algorithm, where we first infer the sparsity pattern of the canonical directions using our fast algorithm, then we shrink each view, i.e. observations of a set of covariates, to contain observations on the sets of covariates selected in the previous step, and compute their canonical directions via any CCA algorithm. We also introduceDirected Sparse CCA, which is able to find associations which are aligned with a specified experiment design, andMulti-View sCCA which is used to discover associations between multiple sets of covariates. Our simulations establish the superior convergence properties and computational efficiency of our algorithm as well as accuracy in terms of the canonical correlation and its ability to recover the supports of the canonical directions. We study the associations between metabolomics, trasncriptomics and microbiomics in a multi-omic study usingMuLe, which is an R-package that implements our approach, in order to form hypotheses on mechanisms of adaptations of Drosophila Melanogaster to high doses of environmental toxicants, specifically Atrazine, which is a commonly used chemical fertilizer.
BLOCCS: Block Sparse Canonical Correlation Analysis With Application To Interpretable Omics Integration
Sep 17, 2019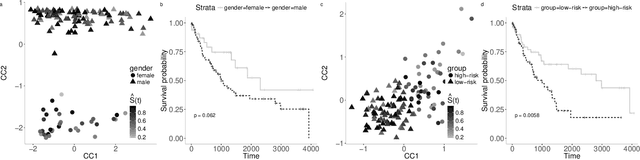
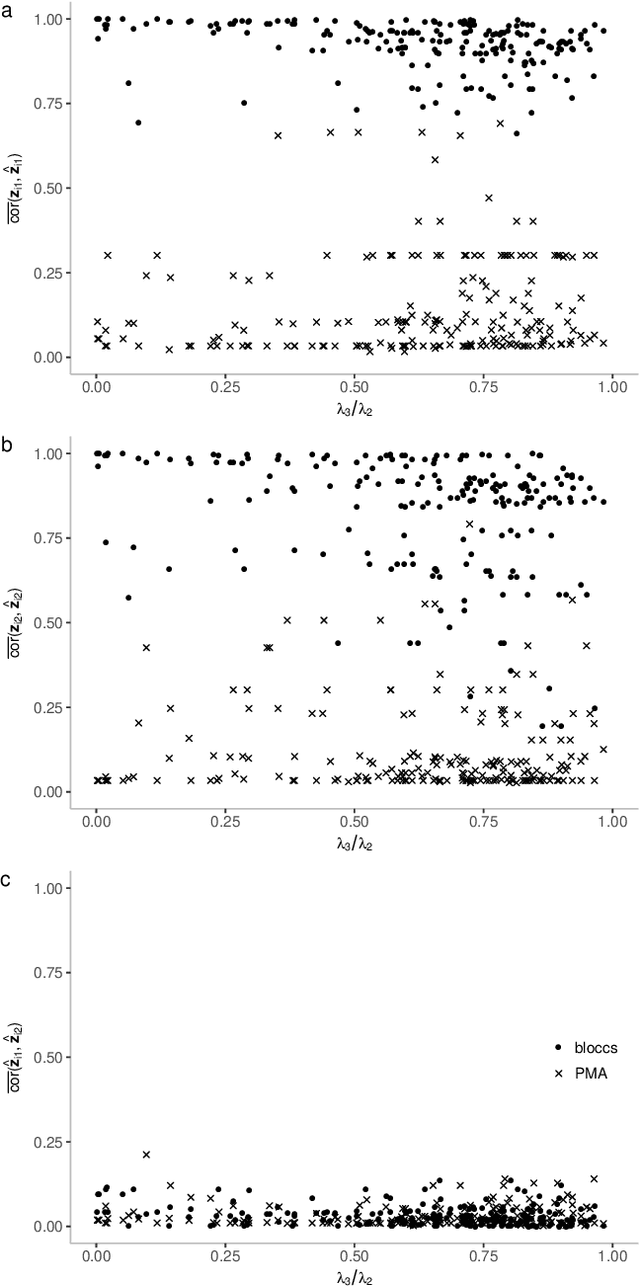
We introduce Block Sparse Canonical Correlation Analysis which estimates multiple pairs of canonical directions (together a "block") at once, resulting in significantly improved orthogonality of the sparse directions which, we demonstrate, translates to more interpretable solutions. Our approach builds on the sparse CCA method of (Solari, Brown, and Bickel 2019) in that we also express the bi-convex objective of our block formulation as a concave minimization problem over an orthogonal k-frame in a unit Euclidean ball, which in turn, due to concavity of the objective, is shrunk to a Stiefel manifold, which is optimized via gradient descent algorithm. Our simulations show that our method outperforms existing sCCA algorithms and implementations in terms of computational cost and stability, mainly due to the drastic shrinkage of our search space, and the correlation within and orthogonality between pairs of estimated canonical covariates. Finally, we apply our method, available as an R-package called BLOCCS, to multi-omic data on Lung Squamous Cell Carcinoma(LUSC) obtained via The Cancer Genome Atlas, and demonstrate its capability in capturing meaningful biological associations relevant to the hypothesis under study rather than spurious dominant variations.
Refining interaction search through signed iterative Random Forests
Oct 16, 2018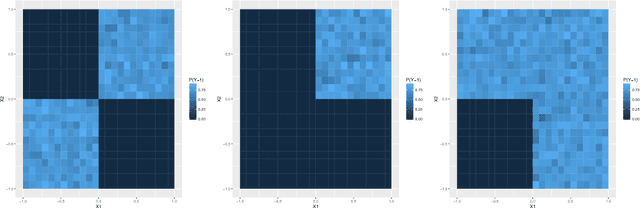
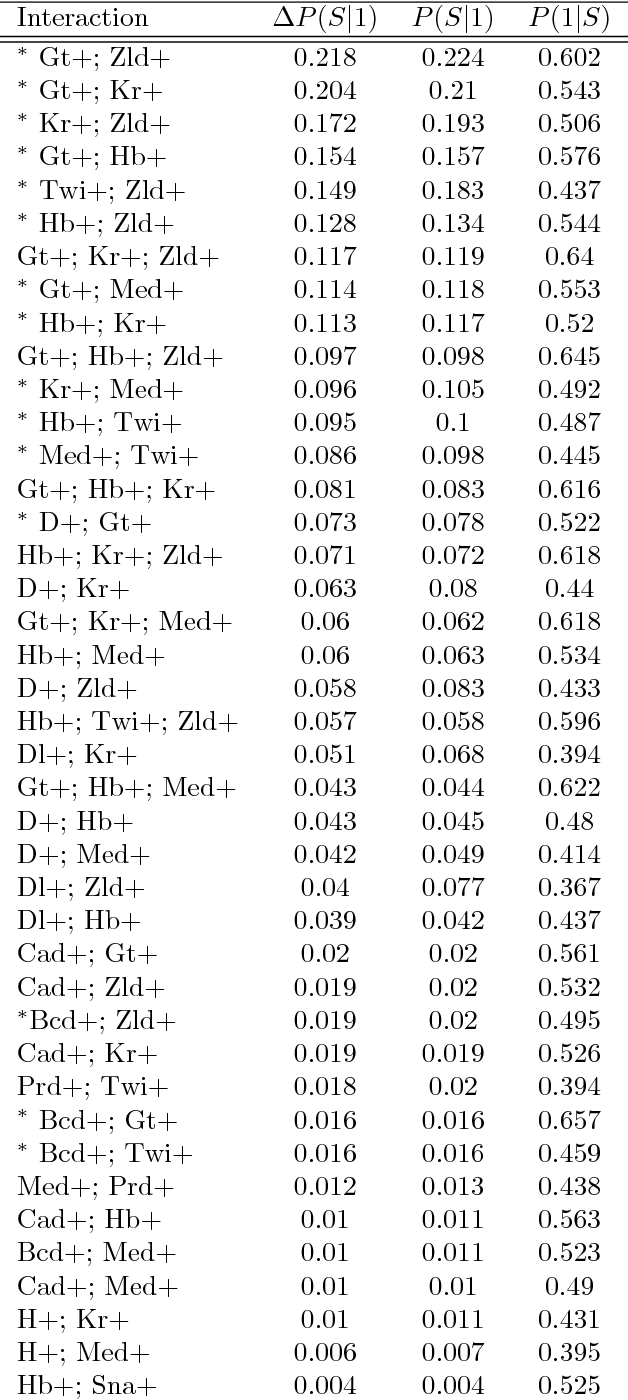
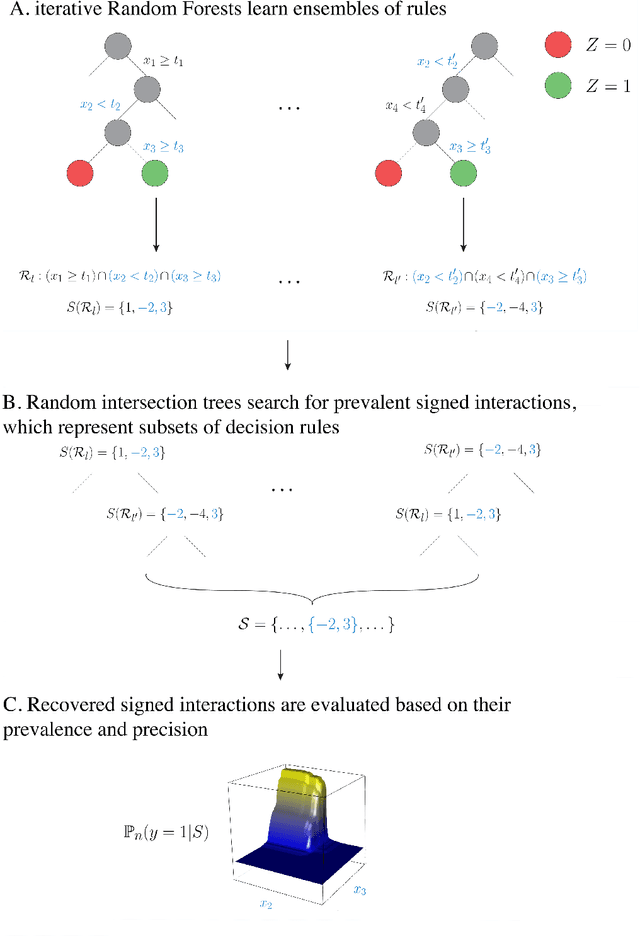
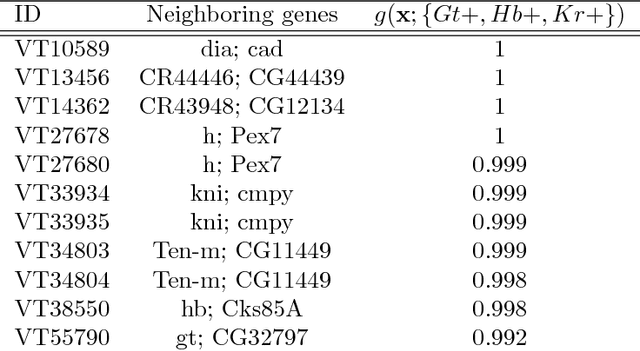
Advances in supervised learning have enabled accurate prediction in biological systems governed by complex interactions among biomolecules. However, state-of-the-art predictive algorithms are typically black-boxes, learning statistical interactions that are difficult to translate into testable hypotheses. The iterative Random Forest algorithm took a step towards bridging this gap by providing a computationally tractable procedure to identify the stable, high-order feature interactions that drive the predictive accuracy of Random Forests (RF). Here we refine the interactions identified by iRF to explicitly map responses as a function of interacting features. Our method, signed iRF, describes subsets of rules that frequently occur on RF decision paths. We refer to these rule subsets as signed interactions. Signed interactions share not only the same set of interacting features but also exhibit similar thresholding behavior, and thus describe a consistent functional relationship between interacting features and responses. We describe stable and predictive importance metrics to rank signed interactions. For each SPIM, we define null importance metrics that characterize its expected behavior under known structure. We evaluate our proposed approach in biologically inspired simulations and two case studies: predicting enhancer activity and spatial gene expression patterns. In the case of enhancer activity, s-iRF recovers one of the few experimentally validated high-order interactions and suggests novel enhancer elements where this interaction may be active. In the case of spatial gene expression patterns, s-iRF recovers all 11 reported links in the gap gene network. By refining the process of interaction recovery, our approach has the potential to guide mechanistic inquiry into systems whose scale and complexity is beyond human comprehension.
Iterative Random Forests to detect predictive and stable high-order interactions
Dec 23, 2017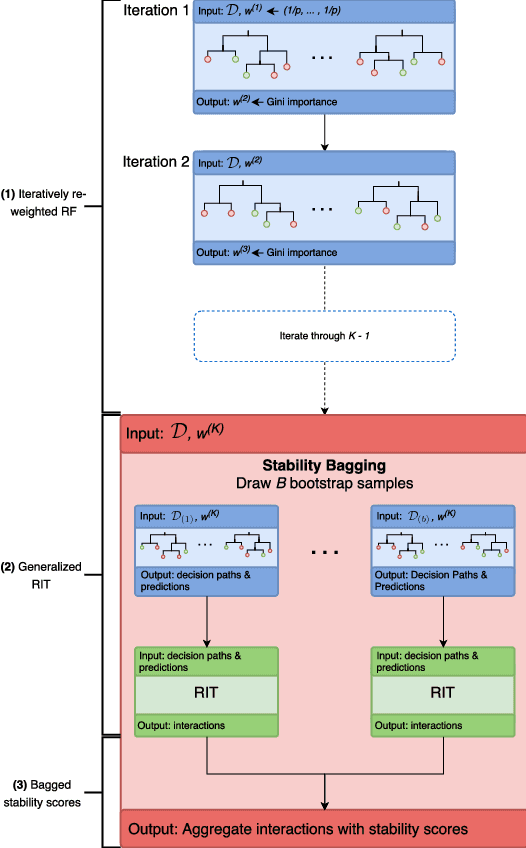
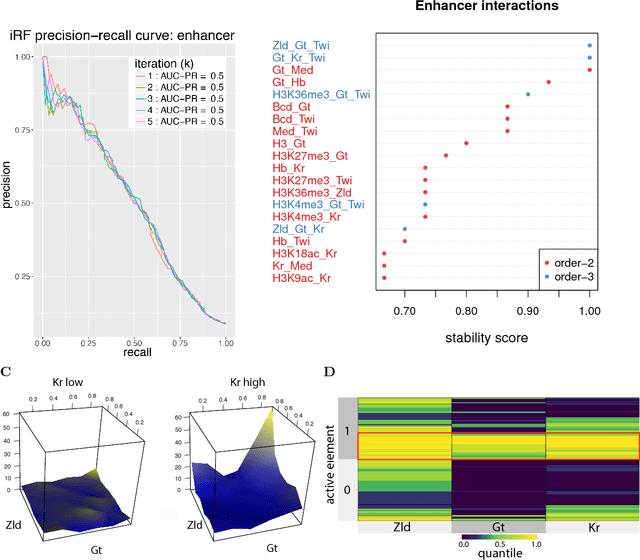
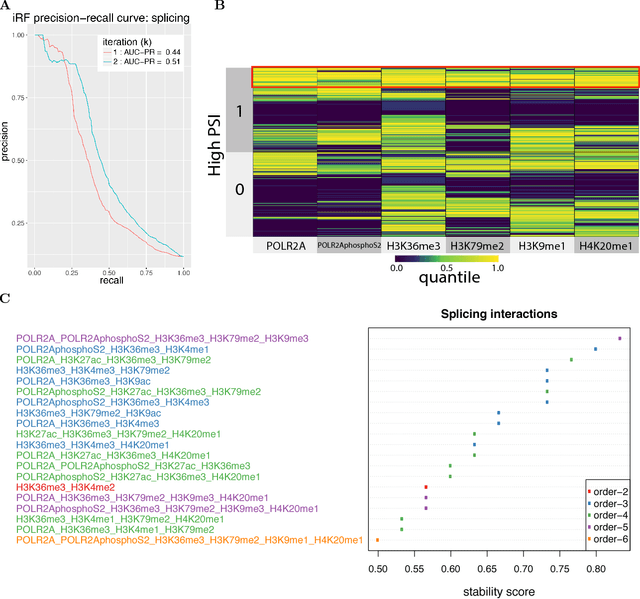
Genomics has revolutionized biology, enabling the interrogation of whole transcriptomes, genome-wide binding sites for proteins, and many other molecular processes. However, individual genomic assays measure elements that interact in vivo as components of larger molecular machines. Understanding how these high-order interactions drive gene expression presents a substantial statistical challenge. Building on Random Forests (RF), Random Intersection Trees (RITs), and through extensive, biologically inspired simulations, we developed the iterative Random Forest algorithm (iRF). iRF trains a feature-weighted ensemble of decision trees to detect stable, high-order interactions with same order of computational cost as RF. We demonstrate the utility of iRF for high-order interaction discovery in two prediction problems: enhancer activity in the early Drosophila embryo and alternative splicing of primary transcripts in human derived cell lines. In Drosophila, among the 20 pairwise transcription factor interactions iRF identifies as stable (returned in more than half of bootstrap replicates), 80% have been previously reported as physical interactions. Moreover, novel third-order interactions, e.g. between Zelda (Zld), Giant (Gt), and Twist (Twi), suggest high-order relationships that are candidates for follow-up experiments. In human-derived cells, iRF re-discovered a central role of H3K36me3 in chromatin-mediated splicing regulation, and identified novel 5th and 6th order interactions, indicative of multi-valent nucleosomes with specific roles in splicing regulation. By decoupling the order of interactions from the computational cost of identification, iRF opens new avenues of inquiry into the molecular mechanisms underlying genome biology.