 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIterative Random Forests to detect predictive and stable high-order interactions
Paper and Code
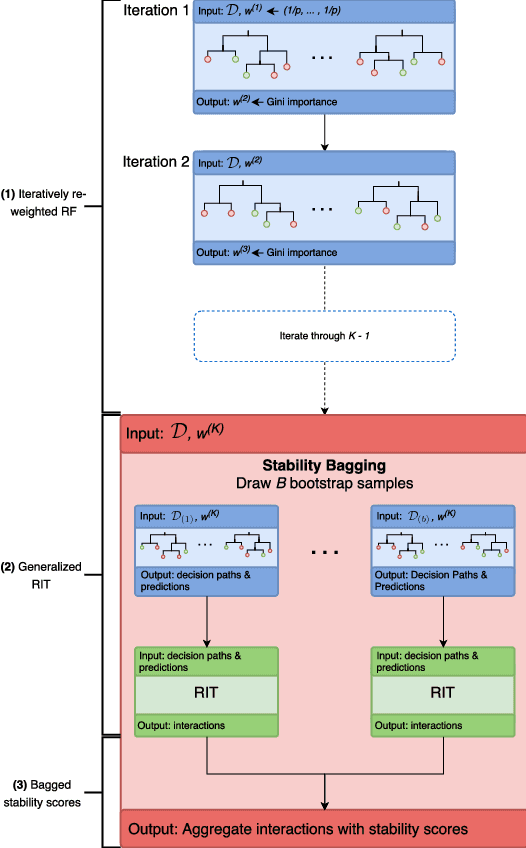
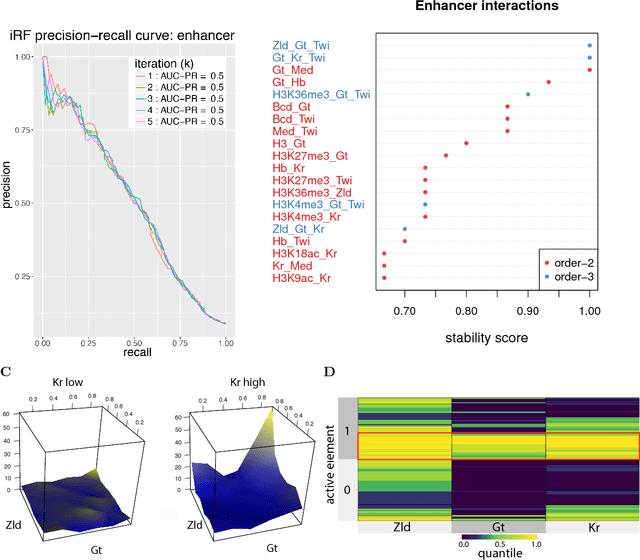
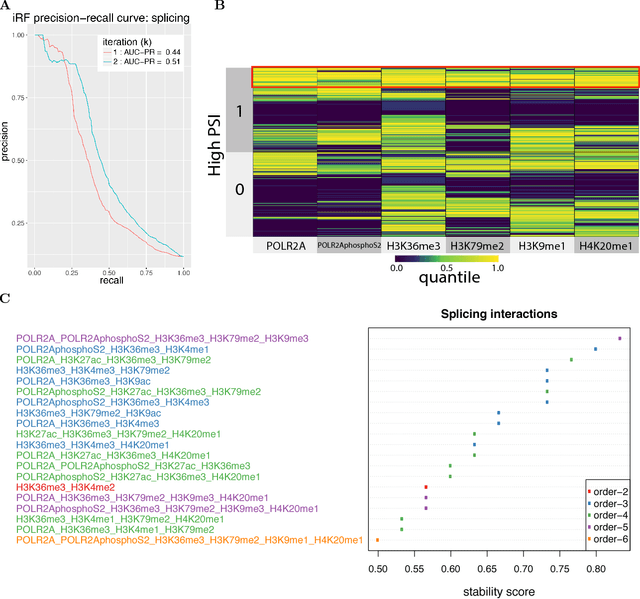
Genomics has revolutionized biology, enabling the interrogation of whole transcriptomes, genome-wide binding sites for proteins, and many other molecular processes. However, individual genomic assays measure elements that interact in vivo as components of larger molecular machines. Understanding how these high-order interactions drive gene expression presents a substantial statistical challenge. Building on Random Forests (RF), Random Intersection Trees (RITs), and through extensive, biologically inspired simulations, we developed the iterative Random Forest algorithm (iRF). iRF trains a feature-weighted ensemble of decision trees to detect stable, high-order interactions with same order of computational cost as RF. We demonstrate the utility of iRF for high-order interaction discovery in two prediction problems: enhancer activity in the early Drosophila embryo and alternative splicing of primary transcripts in human derived cell lines. In Drosophila, among the 20 pairwise transcription factor interactions iRF identifies as stable (returned in more than half of bootstrap replicates), 80% have been previously reported as physical interactions. Moreover, novel third-order interactions, e.g. between Zelda (Zld), Giant (Gt), and Twist (Twi), suggest high-order relationships that are candidates for follow-up experiments. In human-derived cells, iRF re-discovered a central role of H3K36me3 in chromatin-mediated splicing regulation, and identified novel 5th and 6th order interactions, indicative of multi-valent nucleosomes with specific roles in splicing regulation. By decoupling the order of interactions from the computational cost of identification, iRF opens new avenues of inquiry into the molecular mechanisms underlying genome biology.