 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRefining interaction search through signed iterative Random Forests
Paper and Code
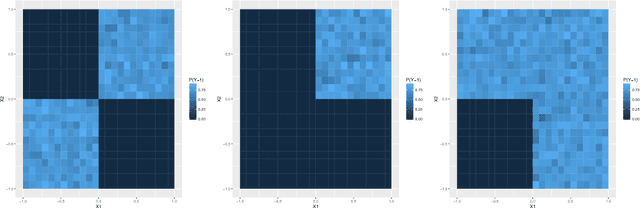
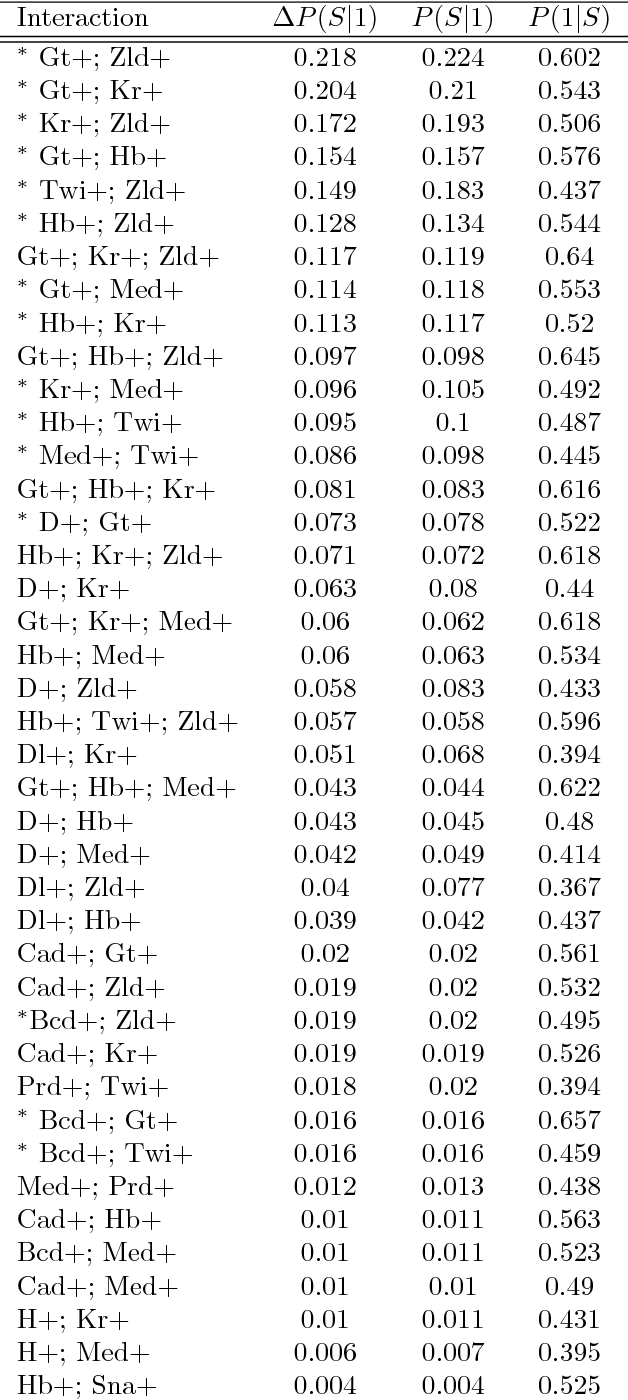
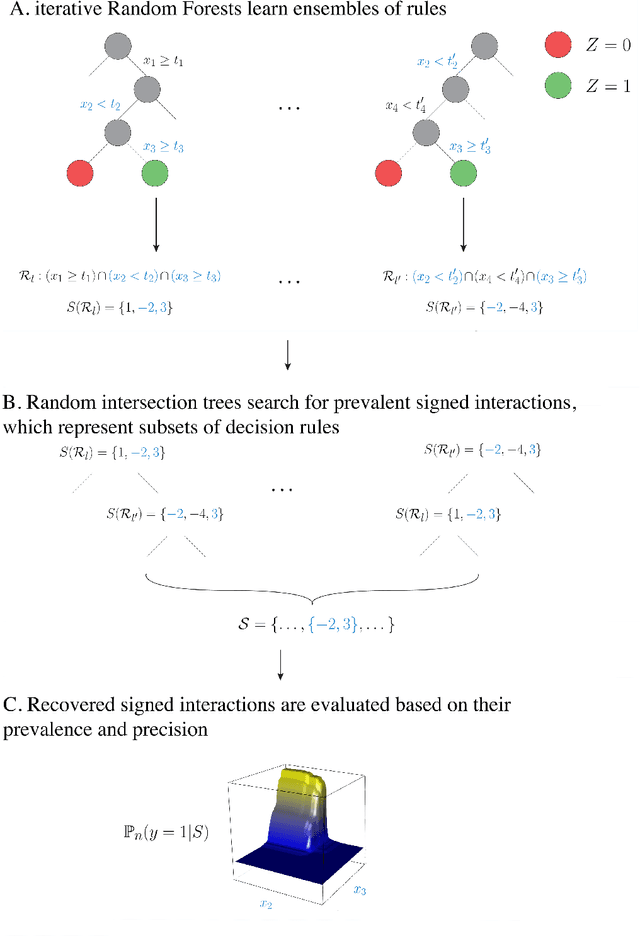
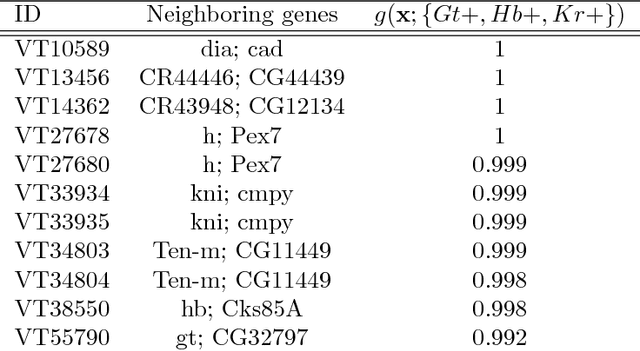
Advances in supervised learning have enabled accurate prediction in biological systems governed by complex interactions among biomolecules. However, state-of-the-art predictive algorithms are typically black-boxes, learning statistical interactions that are difficult to translate into testable hypotheses. The iterative Random Forest algorithm took a step towards bridging this gap by providing a computationally tractable procedure to identify the stable, high-order feature interactions that drive the predictive accuracy of Random Forests (RF). Here we refine the interactions identified by iRF to explicitly map responses as a function of interacting features. Our method, signed iRF, describes subsets of rules that frequently occur on RF decision paths. We refer to these rule subsets as signed interactions. Signed interactions share not only the same set of interacting features but also exhibit similar thresholding behavior, and thus describe a consistent functional relationship between interacting features and responses. We describe stable and predictive importance metrics to rank signed interactions. For each SPIM, we define null importance metrics that characterize its expected behavior under known structure. We evaluate our proposed approach in biologically inspired simulations and two case studies: predicting enhancer activity and spatial gene expression patterns. In the case of enhancer activity, s-iRF recovers one of the few experimentally validated high-order interactions and suggests novel enhancer elements where this interaction may be active. In the case of spatial gene expression patterns, s-iRF recovers all 11 reported links in the gap gene network. By refining the process of interaction recovery, our approach has the potential to guide mechanistic inquiry into systems whose scale and complexity is beyond human comprehension.