 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeExhaustive Exploitation of Nature-inspired Computation for Cancer Screening in an Ensemble Manner
Apr 06, 2024



Accurate screening of cancer types is crucial for effective cancer detection and precise treatment selection. However, the association between gene expression profiles and tumors is often limited to a small number of biomarker genes. While computational methods using nature-inspired algorithms have shown promise in selecting predictive genes, existing techniques are limited by inefficient search and poor generalization across diverse datasets. This study presents a framework termed Evolutionary Optimized Diverse Ensemble Learning (EODE) to improve ensemble learning for cancer classification from gene expression data. The EODE methodology combines an intelligent grey wolf optimization algorithm for selective feature space reduction, guided random injection modeling for ensemble diversity enhancement, and subset model optimization for synergistic classifier combinations. Extensive experiments were conducted across 35 gene expression benchmark datasets encompassing varied cancer types. Results demonstrated that EODE obtained significantly improved screening accuracy over individual and conventionally aggregated models. The integrated optimization of advanced feature selection, directed specialized modeling, and cooperative classifier ensembles helps address key challenges in current nature-inspired approaches. This provides an effective framework for robust and generalized ensemble learning with gene expression biomarkers. Specifically, we have opened EODE source code on Github at https://github.com/wangxb96/EODE.
DiffDTM: A conditional structure-free framework for bioactive molecules generation targeted for dual proteins
Jun 24, 2023Advances in deep generative models shed light on de novo molecule generation with desired properties. However, molecule generation targeted for dual protein targets still faces formidable challenges including protein 3D structure data requisition for model training, auto-regressive sampling, and model generalization for unseen targets. Here, we proposed DiffDTM, a novel conditional structure-free deep generative model based on a diffusion model for dual targets based molecule generation to address the above issues. Specifically, DiffDTM receives protein sequences and molecular graphs as inputs instead of protein and molecular conformations and incorporates an information fusion module to achieve conditional generation in a one-shot manner. We have conducted comprehensive multi-view experiments to demonstrate that DiffDTM can generate drug-like, synthesis-accessible, novel, and high-binding affinity molecules targeting specific dual proteins, outperforming the state-of-the-art (SOTA) models in terms of multiple evaluation metrics. Furthermore, we utilized DiffDTM to generate molecules towards dopamine receptor D2 and 5-hydroxytryptamine receptor 1A as new antipsychotics. The experimental results indicate that DiffDTM can be easily plugged into unseen dual targets to generate bioactive molecules, addressing the issues of requiring insufficient active molecule data for training as well as the need to retrain when encountering new targets.
MDM: Molecular Diffusion Model for 3D Molecule Generation
Sep 13, 2022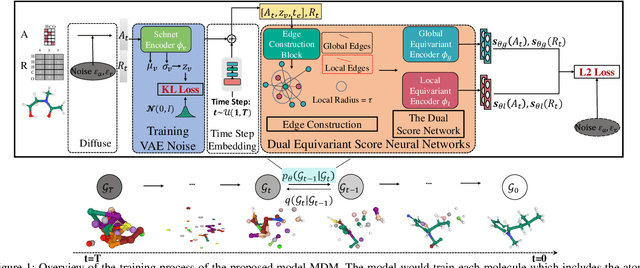
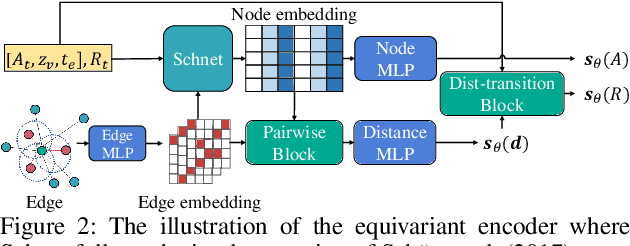
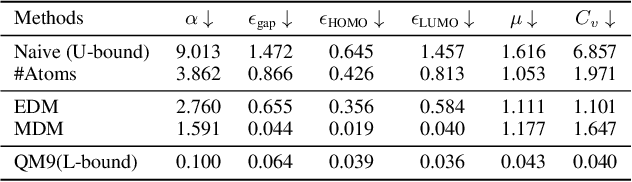
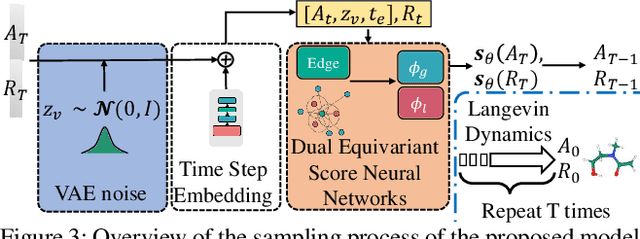
Molecule generation, especially generating 3D molecular geometries from scratch (i.e., 3D \textit{de novo} generation), has become a fundamental task in drug designs. Existing diffusion-based 3D molecule generation methods could suffer from unsatisfactory performances, especially when generating large molecules. At the same time, the generated molecules lack enough diversity. This paper proposes a novel diffusion model to address those two challenges. First, interatomic relations are not in molecules' 3D point cloud representations. Thus, it is difficult for existing generative models to capture the potential interatomic forces and abundant local constraints. To tackle this challenge, we propose to augment the potential interatomic forces and further involve dual equivariant encoders to encode interatomic forces of different strengths. Second, existing diffusion-based models essentially shift elements in geometry along the gradient of data density. Such a process lacks enough exploration in the intermediate steps of the Langevin dynamics. To address this issue, we introduce a distributional controlling variable in each diffusion/reverse step to enforce thorough explorations and further improve generation diversity. Extensive experiments on multiple benchmarks demonstrate that the proposed model significantly outperforms existing methods for both unconditional and conditional generation tasks. We also conduct case studies to help understand the physicochemical properties of the generated molecules.
A Self-adaptive Weighted Differential Evolution Approach for Large-scale Feature Selection
Oct 27, 2021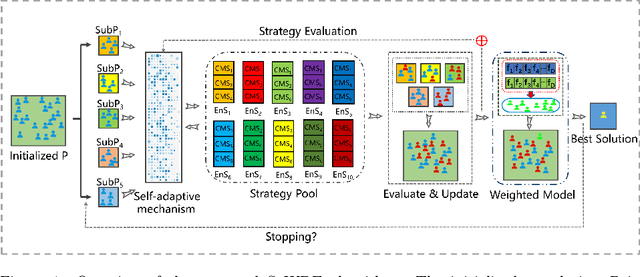

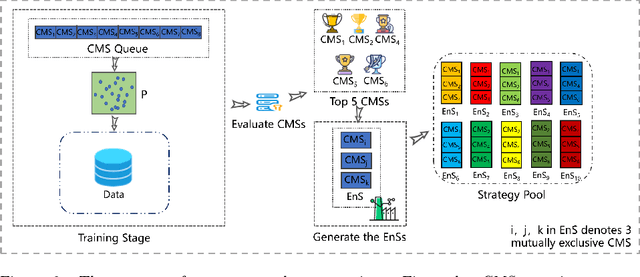

Recently, many evolutionary computation methods have been developed to solve the feature selection problem. However, the studies focused mainly on small-scale issues, resulting in stagnation issues in local optima and numerical instability when dealing with large-scale feature selection dilemmas. To address these challenges, this paper proposes a novel weighted differential evolution algorithm based on self-adaptive mechanism, named SaWDE, to solve large-scale feature selection. First, a multi-population mechanism is adopted to enhance the diversity of the population. Then, we propose a new self-adaptive mechanism that selects several strategies from a strategy pool to capture the diverse characteristics of the datasets from the historical information. Finally, a weighted model is designed to identify the important features, which enables our model to generate the most suitable feature-selection solution. We demonstrate the effectiveness of our algorithm on twelve large-scale datasets. The performance of SaWDE is superior compared to six non-EC algorithms and six other EC algorithms, on both training and test datasets and on subset size, indicating that our algorithm is a favorable tool to solve the large-scale feature selection problem. Moreover, we have experimented SaWDE with six EC algorithms on twelve higher-dimensional data, which demonstrates that SaWDE is more robust and efficient compared to those state-of-the-art methods. SaWDE source code is available on Github at https://github.com/wangxb96/SaWDE.
EGFI: Drug-Drug Interaction Extraction and Generation with Fusion of Enriched Entity and Sentence Information
Jan 25, 2021
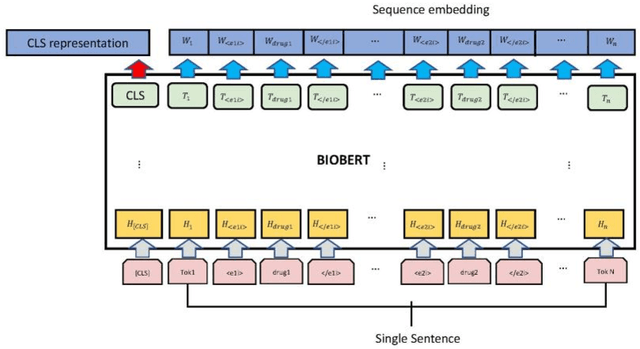
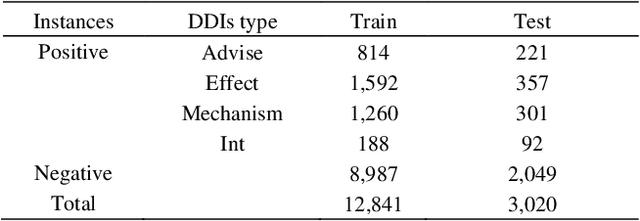
The rapid growth in literature accumulates diverse and yet comprehensive biomedical knowledge hidden to be mined such as drug interactions. However, it is difficult to extract the heterogeneous knowledge to retrieve or even discover the latest and novel knowledge in an efficient manner. To address such a problem, we propose EGFI for extracting and consolidating drug interactions from large-scale medical literature text data. Specifically, EGFI consists of two parts: classification and generation. In the classification part, EGFI encompasses the language model BioBERT which has been comprehensively pre-trained on biomedical corpus. In particular, we propose the multi-head attention mechanism and pack BiGRU to fuse multiple semantic information for rigorous context modeling. In the generation part, EGFI utilizes another pre-trained language model BioGPT-2 where the generation sentences are selected based on filtering rules. We evaluated the classification part on "DDIs 2013" dataset and "DTIs" dataset, achieving the FI score of 0.842 and 0.720 respectively. Moreover, we applied the classification part to distinguish high-quality generated sentences and verified with the exiting growth truth to confirm the filtered sentences. The generated sentences that are not recorded in DrugBank and DDIs 2013 dataset also demonstrate the potential of EGFI to identify novel drug relationships.
Review of Single-cell RNA-seq Data Clustering for Cell Type Identification and Characterization
Jan 03, 2020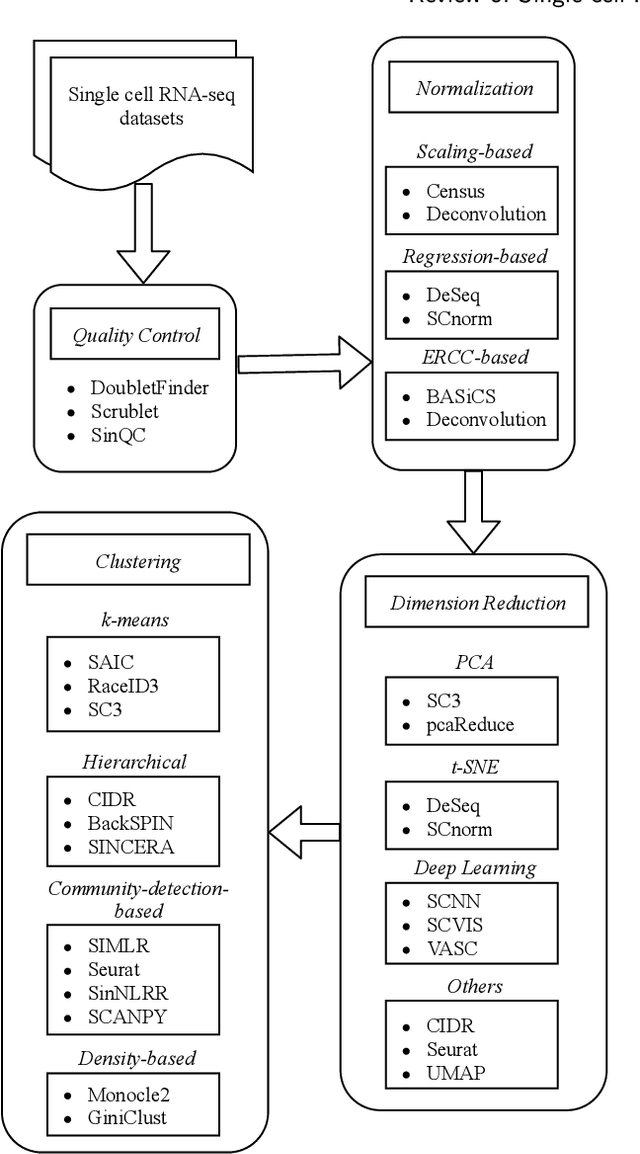
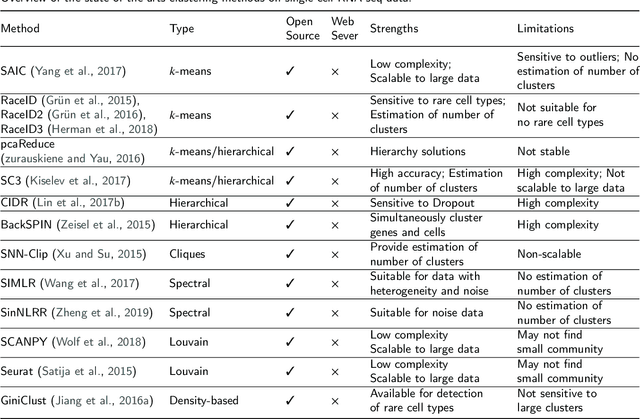
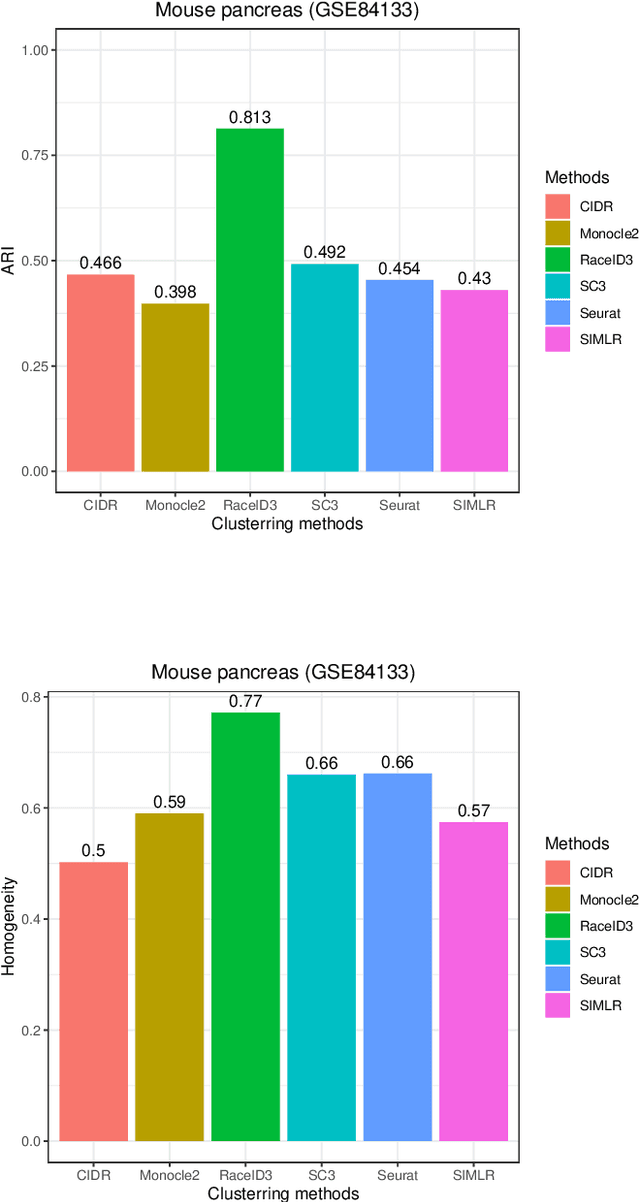

In recent years, the advances in single-cell RNA-seq techniques have enabled us to perform large-scale transcriptomic profiling at single-cell resolution in a high-throughput manner. Unsupervised learning such as data clustering has become the central component to identify and characterize novel cell types and gene expression patterns. In this study, we review the existing single-cell RNA-seq data clustering methods with critical insights into the related advantages and limitations. In addition, we also review the upstream single-cell RNA-seq data processing techniques such as quality control, normalization, and dimension reduction. We conduct performance comparison experiments to evaluate several popular single-cell RNA-seq clustering approaches on two single-cell transcriptomic datasets.
Context awareness and embedding for biomedical event extraction
May 02, 2019
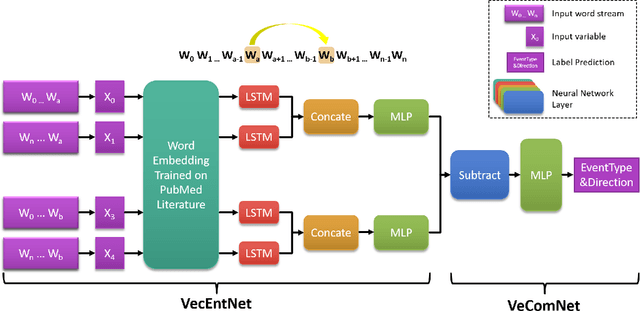
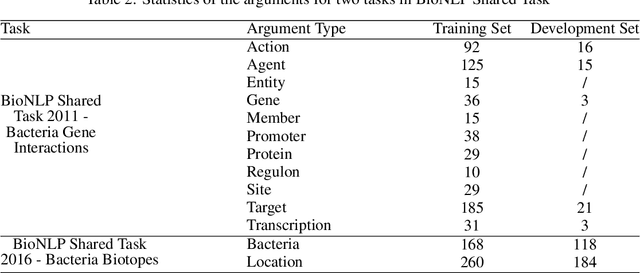
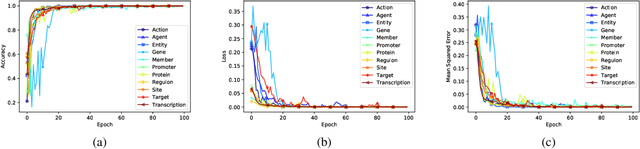
Motivation: Biomedical event detection is fundamental for information extraction in molecular biology and biomedical research. The detected events form the central basis for comprehensive biomedical knowledge fusion, facilitating the digestion of massive information influx from literature. Limited by the feature context, the existing event detection models are mostly applicable for a single task. A general and scalable computational model is desiderated for biomedical knowledge management. Results: We consider and propose a bottom-up detection framework to identify the events from recognized arguments. To capture the relations between the arguments, we trained a bi-directional Long Short-Term Memory (LSTM) network to model their context embedding. Leveraging the compositional attributes, we further derived the candidate samples for training event classifiers. We built our models on the datasets from BioNLP Shared Task for evaluations. Our method achieved the average F-scores of 0.81 and 0.92 on BioNLPST-BGI and BioNLPST-BB datasets respectively. Comparing with 7 state-of-the-art methods, our method nearly doubled the existing F-score performance (0.92 vs 0.56) on the BioNLPST-BB dataset. Case studies were conducted to reveal the underlying reasons. Availability: https://github.com/cskyan/evntextrc
A Short Survey on Data Clustering Algorithms
Nov 25, 2015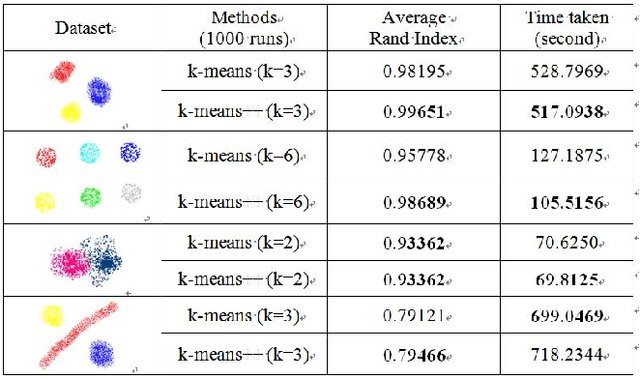
With rapidly increasing data, clustering algorithms are important tools for data analytics in modern research. They have been successfully applied to a wide range of domains; for instance, bioinformatics, speech recognition, and financial analysis. Formally speaking, given a set of data instances, a clustering algorithm is expected to divide the set of data instances into the subsets which maximize the intra-subset similarity and inter-subset dissimilarity, where a similarity measure is defined beforehand. In this work, the state-of-the-arts clustering algorithms are reviewed from design concept to methodology; Different clustering paradigms are discussed. Advanced clustering algorithms are also discussed. After that, the existing clustering evaluation metrics are reviewed. A summary with future insights is provided at the end.
Evolutionary Algorithms: Concepts, Designs, and Applications in Bioinformatics: Evolutionary Algorithms for Bioinformatics
Aug 03, 2015
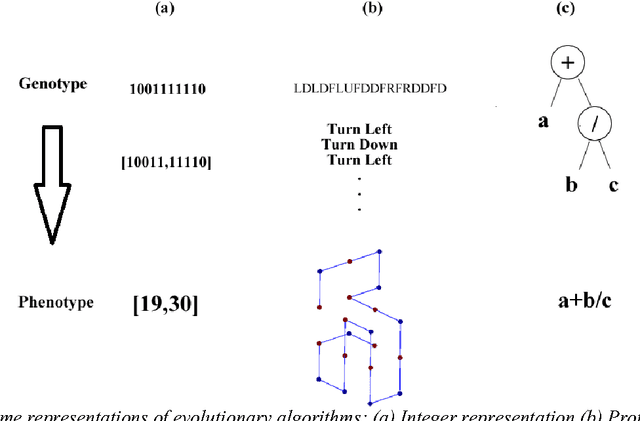
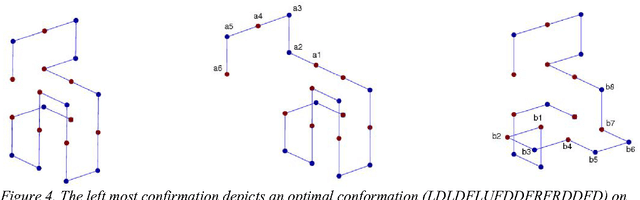
Since genetic algorithm was proposed by John Holland (Holland J. H., 1975) in the early 1970s, the study of evolutionary algorithm has emerged as a popular research field (Civicioglu & Besdok, 2013). Researchers from various scientific and engineering disciplines have been digging into this field, exploring the unique power of evolutionary algorithms (Hadka & Reed, 2013). Many applications have been successfully proposed in the past twenty years. For example, mechanical design (Lampinen & Zelinka, 1999), electromagnetic optimization (Rahmat-Samii & Michielssen, 1999), environmental protection (Bertini, Felice, Moretti, & Pizzuti, 2010), finance (Larkin & Ryan, 2010), musical orchestration (Esling, Carpentier, & Agon, 2010), pipe routing (Furuholmen, Glette, Hovin, & Torresen, 2010), and nuclear reactor core design (Sacco, Henderson, Rios-Coelho, Ali, & Pereira, 2009). In particular, its function optimization capability was highlighted (Goldberg & Richardson, 1987) because of its high adaptability to different function landscapes, to which we cannot apply traditional optimization techniques (Wong, Leung, & Wong, 2009). Here we review the applications of evolutionary algorithms in bioinformatics.
Unsupervised Learning in Genome Informatics
Aug 03, 2015

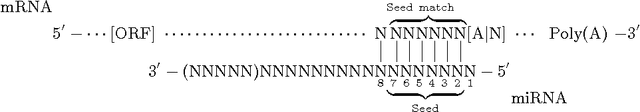
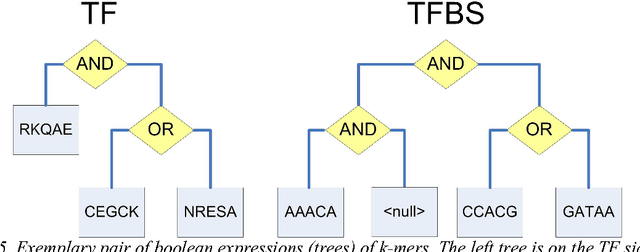
With different genomes available, unsupervised learning algorithms are essential in learning genome-wide biological insights. Especially, the functional characterization of different genomes is essential for us to understand lives. In this book chapter, we review the state-of-the-art unsupervised learning algorithms for genome informatics from DNA to MicroRNA. DNA (DeoxyriboNucleic Acid) is the basic component of genomes. A significant fraction of DNA regions (transcription factor binding sites) are bound by proteins (transcription factors) to regulate gene expression at different development stages in different tissues. To fully understand genetics, it is necessary of us to apply unsupervised learning algorithms to learn and infer those DNA regions. Here we review several unsupervised learning methods for deciphering the genome-wide patterns of those DNA regions. MicroRNA (miRNA), a class of small endogenous non-coding RNA (RiboNucleic acid) species, regulate gene expression post-transcriptionally by forming imperfect base-pair with the target sites primarily at the 3$'$ untranslated regions of the messenger RNAs. Since the 1993 discovery of the first miRNA \emph{let-7} in worms, a vast amount of studies have been dedicated to functionally characterizing the functional impacts of miRNA in a network context to understand complex diseases such as cancer. Here we review several representative unsupervised learning frameworks on inferring miRNA regulatory network by exploiting the static sequence-based information pertinent to the prior knowledge of miRNA targeting and the dynamic information of miRNA activities implicated by the recently available large data compendia, which interrogate genome-wide expression profiles of miRNAs and/or mRNAs across various cell conditions.