 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeExhaustive Exploitation of Nature-inspired Computation for Cancer Screening in an Ensemble Manner
Apr 06, 2024



Accurate screening of cancer types is crucial for effective cancer detection and precise treatment selection. However, the association between gene expression profiles and tumors is often limited to a small number of biomarker genes. While computational methods using nature-inspired algorithms have shown promise in selecting predictive genes, existing techniques are limited by inefficient search and poor generalization across diverse datasets. This study presents a framework termed Evolutionary Optimized Diverse Ensemble Learning (EODE) to improve ensemble learning for cancer classification from gene expression data. The EODE methodology combines an intelligent grey wolf optimization algorithm for selective feature space reduction, guided random injection modeling for ensemble diversity enhancement, and subset model optimization for synergistic classifier combinations. Extensive experiments were conducted across 35 gene expression benchmark datasets encompassing varied cancer types. Results demonstrated that EODE obtained significantly improved screening accuracy over individual and conventionally aggregated models. The integrated optimization of advanced feature selection, directed specialized modeling, and cooperative classifier ensembles helps address key challenges in current nature-inspired approaches. This provides an effective framework for robust and generalized ensemble learning with gene expression biomarkers. Specifically, we have opened EODE source code on Github at https://github.com/wangxb96/EODE.
A Self-adaptive Weighted Differential Evolution Approach for Large-scale Feature Selection
Oct 27, 2021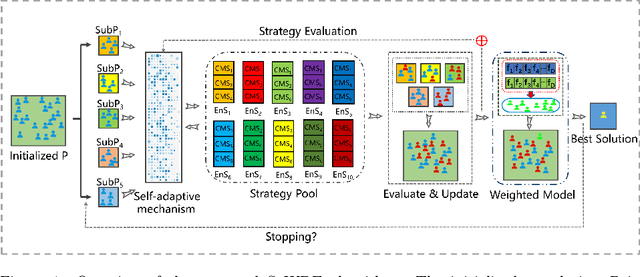

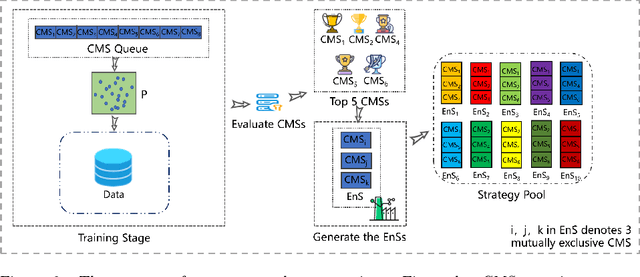

Recently, many evolutionary computation methods have been developed to solve the feature selection problem. However, the studies focused mainly on small-scale issues, resulting in stagnation issues in local optima and numerical instability when dealing with large-scale feature selection dilemmas. To address these challenges, this paper proposes a novel weighted differential evolution algorithm based on self-adaptive mechanism, named SaWDE, to solve large-scale feature selection. First, a multi-population mechanism is adopted to enhance the diversity of the population. Then, we propose a new self-adaptive mechanism that selects several strategies from a strategy pool to capture the diverse characteristics of the datasets from the historical information. Finally, a weighted model is designed to identify the important features, which enables our model to generate the most suitable feature-selection solution. We demonstrate the effectiveness of our algorithm on twelve large-scale datasets. The performance of SaWDE is superior compared to six non-EC algorithms and six other EC algorithms, on both training and test datasets and on subset size, indicating that our algorithm is a favorable tool to solve the large-scale feature selection problem. Moreover, we have experimented SaWDE with six EC algorithms on twelve higher-dimensional data, which demonstrates that SaWDE is more robust and efficient compared to those state-of-the-art methods. SaWDE source code is available on Github at https://github.com/wangxb96/SaWDE.
EGFI: Drug-Drug Interaction Extraction and Generation with Fusion of Enriched Entity and Sentence Information
Jan 25, 2021
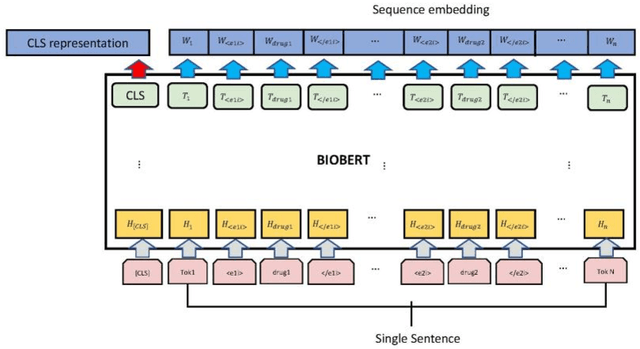
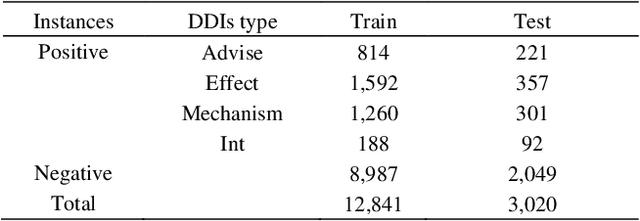
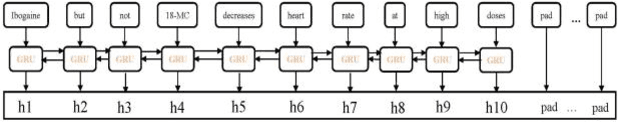
The rapid growth in literature accumulates diverse and yet comprehensive biomedical knowledge hidden to be mined such as drug interactions. However, it is difficult to extract the heterogeneous knowledge to retrieve or even discover the latest and novel knowledge in an efficient manner. To address such a problem, we propose EGFI for extracting and consolidating drug interactions from large-scale medical literature text data. Specifically, EGFI consists of two parts: classification and generation. In the classification part, EGFI encompasses the language model BioBERT which has been comprehensively pre-trained on biomedical corpus. In particular, we propose the multi-head attention mechanism and pack BiGRU to fuse multiple semantic information for rigorous context modeling. In the generation part, EGFI utilizes another pre-trained language model BioGPT-2 where the generation sentences are selected based on filtering rules. We evaluated the classification part on "DDIs 2013" dataset and "DTIs" dataset, achieving the FI score of 0.842 and 0.720 respectively. Moreover, we applied the classification part to distinguish high-quality generated sentences and verified with the exiting growth truth to confirm the filtered sentences. The generated sentences that are not recorded in DrugBank and DDIs 2013 dataset also demonstrate the potential of EGFI to identify novel drug relationships.
Review of Single-cell RNA-seq Data Clustering for Cell Type Identification and Characterization
Jan 03, 2020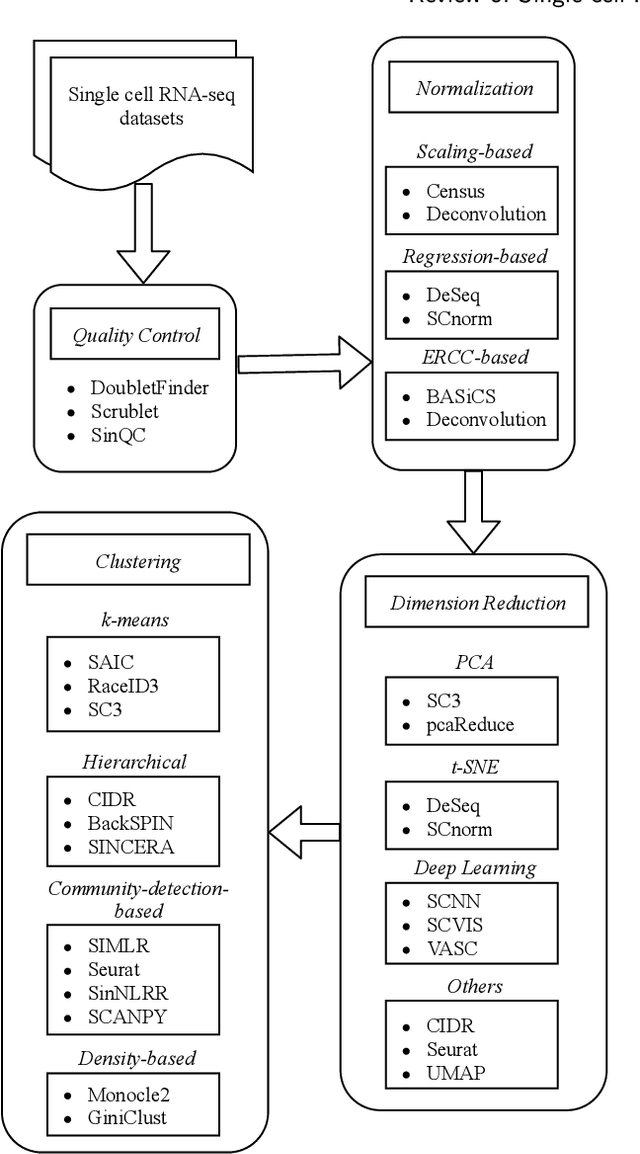
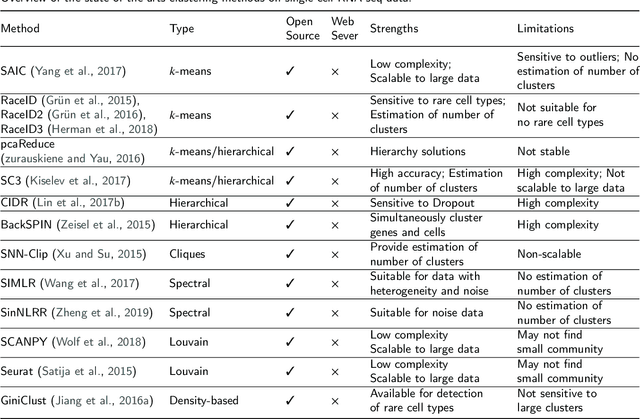
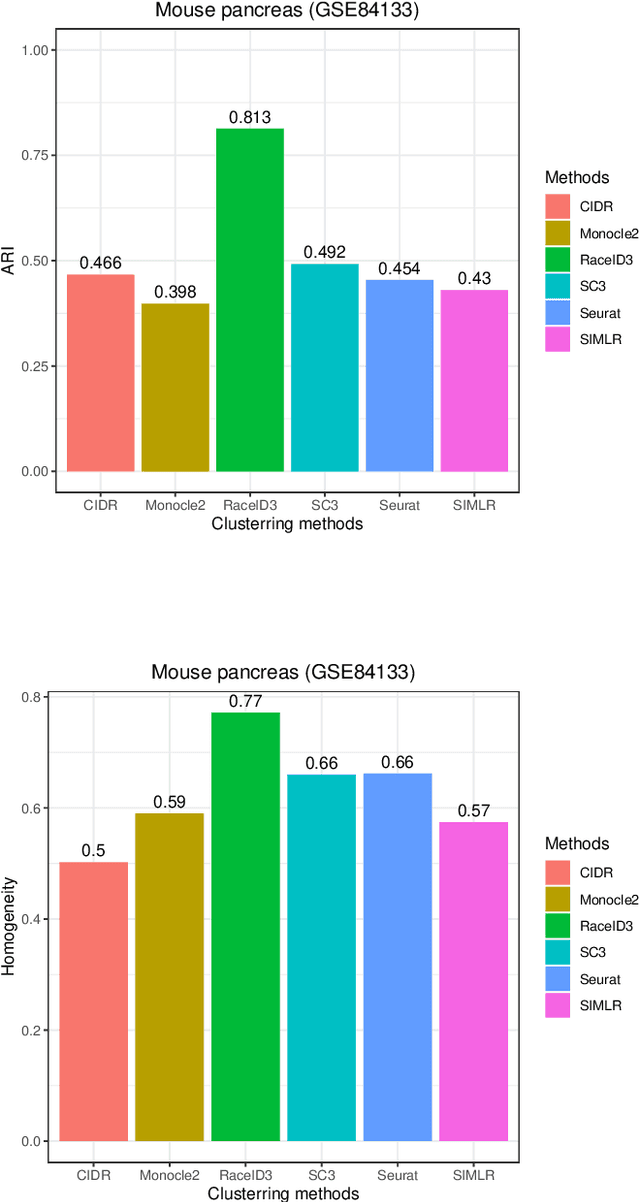

In recent years, the advances in single-cell RNA-seq techniques have enabled us to perform large-scale transcriptomic profiling at single-cell resolution in a high-throughput manner. Unsupervised learning such as data clustering has become the central component to identify and characterize novel cell types and gene expression patterns. In this study, we review the existing single-cell RNA-seq data clustering methods with critical insights into the related advantages and limitations. In addition, we also review the upstream single-cell RNA-seq data processing techniques such as quality control, normalization, and dimension reduction. We conduct performance comparison experiments to evaluate several popular single-cell RNA-seq clustering approaches on two single-cell transcriptomic datasets.