 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Survey on Multimodal Music Emotion Recognition
Apr 26, 2025Multimodal music emotion recognition (MMER) is an emerging discipline in music information retrieval that has experienced a surge in interest in recent years. This survey provides a comprehensive overview of the current state-of-the-art in MMER. Discussing the different approaches and techniques used in this field, the paper introduces a four-stage MMER framework, including multimodal data selection, feature extraction, feature processing, and final emotion prediction. The survey further reveals significant advancements in deep learning methods and the increasing importance of feature fusion techniques. Despite these advancements, challenges such as the need for large annotated datasets, datasets with more modalities, and real-time processing capabilities remain. This paper also contributes to the field by identifying critical gaps in current research and suggesting potential directions for future research. The gaps underscore the importance of developing robust, scalable, a interpretable models for MMER, with implications for applications in music recommendation systems, therapeutic tools, and entertainment.
Leveraging Vision-Language Embeddings for Zero-Shot Learning in Histopathology Images
Mar 13, 2025Zero-shot learning holds tremendous potential for histopathology image analysis by enabling models to generalize to unseen classes without extensive labeled data. Recent advancements in vision-language models (VLMs) have expanded the capabilities of ZSL, allowing models to perform tasks without task-specific fine-tuning. However, applying VLMs to histopathology presents considerable challenges due to the complexity of histopathological imagery and the nuanced nature of diagnostic tasks. In this paper, we propose a novel framework called Multi-Resolution Prompt-guided Hybrid Embedding (MR-PHE) to address these challenges in zero-shot histopathology image classification. MR-PHE leverages multiresolution patch extraction to mimic the diagnostic workflow of pathologists, capturing both fine-grained cellular details and broader tissue structures critical for accurate diagnosis. We introduce a hybrid embedding strategy that integrates global image embeddings with weighted patch embeddings, effectively combining local and global contextual information. Additionally, we develop a comprehensive prompt generation and selection framework, enriching class descriptions with domain-specific synonyms and clinically relevant features to enhance semantic understanding. A similarity-based patch weighting mechanism assigns attention-like weights to patches based on their relevance to class embeddings, emphasizing diagnostically important regions during classification. Our approach utilizes pretrained VLM, CONCH for ZSL without requiring domain-specific fine-tuning, offering scalability and reducing dependence on large annotated datasets. Experimental results demonstrate that MR-PHE not only significantly improves zero-shot classification performance on histopathology datasets but also often surpasses fully supervised models.
GRAPHITE: Graph-Based Interpretable Tissue Examination for Enhanced Explainability in Breast Cancer Histopathology
Jan 08, 2025



Explainable AI (XAI) in medical histopathology is essential for enhancing the interpretability and clinical trustworthiness of deep learning models in cancer diagnosis. However, the black-box nature of these models often limits their clinical adoption. We introduce GRAPHITE (Graph-based Interpretable Tissue Examination), a post-hoc explainable framework designed for breast cancer tissue microarray (TMA) analysis. GRAPHITE employs a multiscale approach, extracting patches at various magnification levels, constructing an hierarchical graph, and utilising graph attention networks (GAT) with scalewise attention (SAN) to capture scale-dependent features. We trained the model on 140 tumour TMA cores and four benign whole slide images from which 140 benign samples were created, and tested it on 53 pathologist-annotated TMA samples. GRAPHITE outperformed traditional XAI methods, achieving a mean average precision (mAP) of 0.56, an area under the receiver operating characteristic curve (AUROC) of 0.94, and a threshold robustness (ThR) of 0.70, indicating that the model maintains high performance across a wide range of thresholds. In clinical utility, GRAPHITE achieved the highest area under the decision curve (AUDC) of 4.17e+5, indicating reliable decision support across thresholds. These results highlight GRAPHITE's potential as a clinically valuable tool in computational pathology, providing interpretable visualisations that align with the pathologists' diagnostic reasoning and support precision medicine.
Attention-Enhanced Lightweight Hourglass Network for Human Pose Estimation
Dec 09, 2024



Pose estimation is a critical task in computer vision with a wide range of applications from activity monitoring to human-robot interaction. However,most of the existing methods are computationally expensive or have complex architecture. Here we propose a lightweight attention based pose estimation network that utilizes depthwise separable convolution and Convolutional Block Attention Module on an hourglass backbone. The network significantly reduces the computational complexity (floating point operations) and the model size (number of parameters) containing only about 10% of parameters of original eight stack Hourglass network.Experiments were conducted on COCO and MPII datasets using a two stack hourglass backbone. The results showed that our model performs well in comparison to six other lightweight pose estimation models with an average precision of 72.07. The model achieves this performance with only 2.3M parameters and 3.7G FLOPs.
TBConvL-Net: A Hybrid Deep Learning Architecture for Robust Medical Image Segmentation
Sep 05, 2024



Deep learning has shown great potential for automated medical image segmentation to improve the precision and speed of disease diagnostics. However, the task presents significant difficulties due to variations in the scale, shape, texture, and contrast of the pathologies. Traditional convolutional neural network (CNN) models have certain limitations when it comes to effectively modelling multiscale context information and facilitating information interaction between skip connections across levels. To overcome these limitations, a novel deep learning architecture is introduced for medical image segmentation, taking advantage of CNNs and vision transformers. Our proposed model, named TBConvL-Net, involves a hybrid network that combines the local features of a CNN encoder-decoder architecture with long-range and temporal dependencies using biconvolutional long-short-term memory (LSTM) networks and vision transformers (ViT). This enables the model to capture contextual channel relationships in the data and account for the uncertainty of segmentation over time. Additionally, we introduce a novel composite loss function that considers both the segmentation robustness and the boundary agreement of the predicted output with the gold standard. Our proposed model shows consistent improvement over the state of the art on ten publicly available datasets of seven different medical imaging modalities.
Deep Joint Denoising and Detection for Enhanced Intracellular Particle Analysis
Aug 15, 2024



Reliable analysis of intracellular dynamic processes in time-lapse fluorescence microscopy images requires complete and accurate tracking of all small particles in all time frames of the image sequences. A fundamental first step towards this goal is particle detection. Given the small size of the particles, their detection is greatly affected by image noise. Recent studies have shown that applying image denoising as a preprocessing step indeed improves particle detection and their subsequent tracking. Deep learning based particle detection methods have shown superior results compared to traditional detection methods. However, they do not explicitly aim to remove noise from the images to facilitate detection. Thus we hypothesize that their performance could be further improved. In this paper, we propose a new deep neural network, called DENODET (denoising-detection network), which performs image denoising and particle detection simultaneously. We show that integrative denoising and detection yields more accurate detection results. Our method achieves superior results compared to state-of-the-art particle detection methods on the particle tracking challenge dataset and our own real fluorescence microscopy image data.
Deep multimodal saliency parcellation of cerebellar pathways: linking microstructure and individual function through explainable multitask learning
Jul 21, 2024



Parcellation of human cerebellar pathways is essential for advancing our understanding of the human brain. Existing diffusion MRI tractography parcellation methods have been successful in defining major cerebellar fibre tracts, while relying solely on fibre tract structure. However, each fibre tract may relay information related to multiple cognitive and motor functions of the cerebellum. Hence, it may be beneficial for parcellation to consider the potential importance of the fibre tracts for individual motor and cognitive functional performance measures. In this work, we propose a multimodal data-driven method for cerebellar pathway parcellation, which incorporates both measures of microstructure and connectivity, and measures of individual functional performance. Our method involves first training a multitask deep network to predict various cognitive and motor measures from a set of fibre tract structural features. The importance of each structural feature for predicting each functional measure is then computed, resulting in a set of structure-function saliency values that are clustered to parcellate cerebellar pathways. We refer to our method as Deep Multimodal Saliency Parcellation (DeepMSP), as it computes the saliency of structural measures for predicting cognitive and motor functional performance, with these saliencies being applied to the task of parcellation. Applying DeepMSP we found that it was feasible to identify multiple cerebellar pathway parcels with unique structure-function saliency patterns that were stable across training folds.
LMBF-Net: A Lightweight Multipath Bidirectional Focal Attention Network for Multifeatures Segmentation
Jul 03, 2024
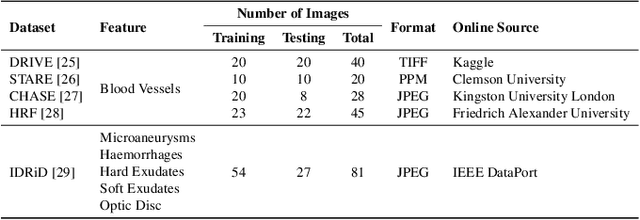
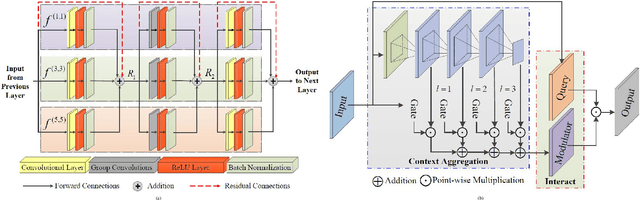
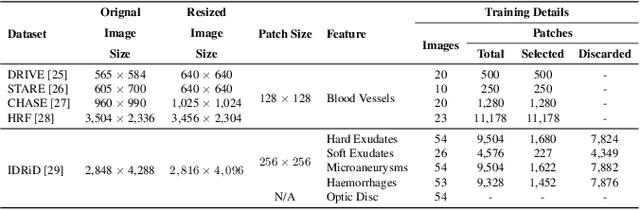
Retinal diseases can cause irreversible vision loss in both eyes if not diagnosed and treated early. Since retinal diseases are so complicated, retinal imaging is likely to show two or more abnormalities. Current deep learning techniques for segmenting retinal images with many labels and attributes have poor detection accuracy and generalisability. This paper presents a multipath convolutional neural network for multifeature segmentation. The proposed network is lightweight and spatially sensitive to information. A patch-based implementation is used to extract local image features, and focal modulation attention blocks are incorporated between the encoder and the decoder for improved segmentation. Filter optimisation is used to prevent filter overlaps and speed up model convergence. A combination of convolution operations and group convolution operations is used to reduce computational costs. This is the first robust and generalisable network capable of segmenting multiple features of fundus images (including retinal vessels, microaneurysms, optic discs, haemorrhages, hard exudates, and soft exudates). The results of our experimental evaluation on more than ten publicly available datasets with multiple features show that the proposed network outperforms recent networks despite having a small number of learnable parameters.
MM-SurvNet: Deep Learning-Based Survival Risk Stratification in Breast Cancer Through Multimodal Data Fusion
Feb 19, 2024Survival risk stratification is an important step in clinical decision making for breast cancer management. We propose a novel deep learning approach for this purpose by integrating histopathological imaging, genetic and clinical data. It employs vision transformers, specifically the MaxViT model, for image feature extraction, and self-attention to capture intricate image relationships at the patient level. A dual cross-attention mechanism fuses these features with genetic data, while clinical data is incorporated at the final layer to enhance predictive accuracy. Experiments on the public TCGA-BRCA dataset show that our model, trained using the negative log likelihood loss function, can achieve superior performance with a mean C-index of 0.64, surpassing existing methods. This advancement facilitates tailored treatment strategies, potentially leading to improved patient outcomes.
BioFusionNet: Deep Learning-Based Survival Risk Stratification in ER+ Breast Cancer Through Multifeature and Multimodal Data Fusion
Feb 16, 2024Breast cancer is a significant health concern affecting millions of women worldwide. Accurate survival risk stratification plays a crucial role in guiding personalised treatment decisions and improving patient outcomes. Here we present BioFusionNet, a deep learning framework that fuses image-derived features with genetic and clinical data to achieve a holistic patient profile and perform survival risk stratification of ER+ breast cancer patients. We employ multiple self-supervised feature extractors, namely DINO and MoCoV3, pretrained on histopathology patches to capture detailed histopathological image features. We then utilise a variational autoencoder (VAE) to fuse these features, and harness the latent space of the VAE to feed into a self-attention network, generating patient-level features. Next, we develop a co-dual-cross-attention mechanism to combine the histopathological features with genetic data, enabling the model to capture the interplay between them. Additionally, clinical data is incorporated using a feed-forward network (FFN), further enhancing predictive performance and achieving comprehensive multimodal feature integration. Furthermore, we introduce a weighted Cox loss function, specifically designed to handle imbalanced survival data, which is a common challenge in the field. The proposed model achieves a mean concordance index (C-index) of 0.77 and a time-dependent area under the curve (AUC) of 0.84, outperforming state-of-the-art methods. It predicts risk (high versus low) with prognostic significance for overall survival (OS) in univariate analysis (HR=2.99, 95% CI: 1.88--4.78, p<0.005), and maintains independent significance in multivariate analysis incorporating standard clinicopathological variables (HR=2.91, 95% CI: 1.80--4.68, p<0.005). The proposed method not only improves model performance but also addresses a critical gap in handling imbalanced data.