 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScaling Atomistic Protein Binder Design with Generative Pretraining and Test-Time Compute
Mar 30, 2026Protein interaction modeling is central to protein design, which has been transformed by machine learning with applications in drug discovery and beyond. In this landscape, structure-based de novo binder design is cast as either conditional generative modeling or sequence optimization via structure predictors ("hallucination"). We argue that this is a false dichotomy and propose Proteina-Complexa, a novel fully atomistic binder generation method unifying both paradigms. We extend recent flow-based latent protein generation architectures and leverage the domain-domain interactions of monomeric computationally predicted protein structures to construct Teddymer, a new large-scale dataset of synthetic binder-target pairs for pretraining. Combined with high-quality experimental multimers, this enables training a strong base model. We then perform inference-time optimization with this generative prior, unifying the strengths of previously distinct generative and hallucination methods. Proteina-Complexa sets a new state of the art in computational binder design benchmarks: it delivers markedly higher in-silico success rates than existing generative approaches, and our novel test-time optimization strategies greatly outperform previous hallucination methods under normalized compute budgets. We also demonstrate interface hydrogen bond optimization, fold class-guided binder generation, and extensions to small molecule targets and enzyme design tasks, again surpassing prior methods. Code, models and new data will be publicly released.
Fold-CP: A Context Parallelism Framework for Biomolecular Modeling
Mar 16, 2026Understanding cellular machinery requires atomic-scale reconstruction of large biomolecular assemblies. However, predicting the structures of these systems has been constrained by hardware memory requirements of models like AlphaFold 3, imposing a practical ceiling of a few thousand residues that can be processed on a single GPU. Here we present NVIDIA BioNeMo Fold-CP, a context parallelism framework that overcomes this barrier by distributing the inference and training pipelines of co-folding models across multiple GPUs. We use the Boltz models as open source reference architectures and implement custom multidimensional primitives that efficiently parallelize both the dense triangular updates and the irregular, data-dependent pattern of window-batched local attention. Our approach achieves efficient memory scaling; for an N-token input distributed across P GPUs, per-device memory scales as $O(N^2/P)$, enabling the structure prediction of assemblies exceeding 30,000 residues on 64 NVIDIA B300 GPUs. We demonstrate the scientific utility of this approach through successful developer use cases: Fold-CP enabled the scoring of over 90% of Comprehensive Resource of Mammalian protein complexes (CORUM) database, as well as folding of disease-relevant PI4KA lipid kinase complex bound to an intrinsically disordered region without cropping. By providing a scalable pathway for modeling massive systems with full global context, Fold-CP represents a significant step toward the realization of a virtual cell.
BioNeMo Framework: a modular, high-performance library for AI model development in drug discovery
Nov 15, 2024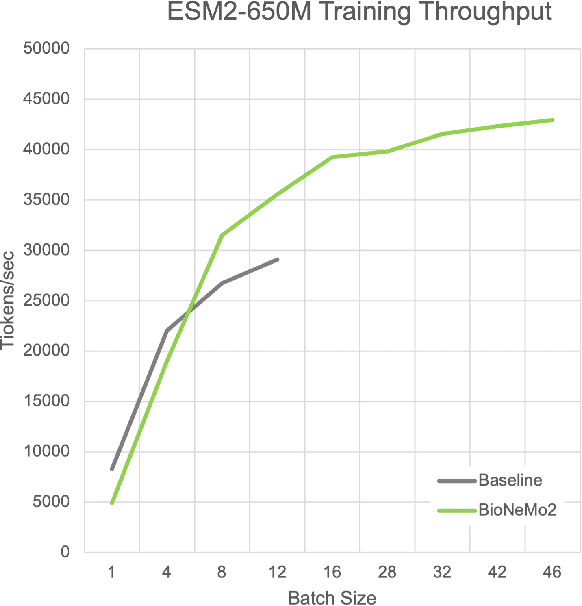
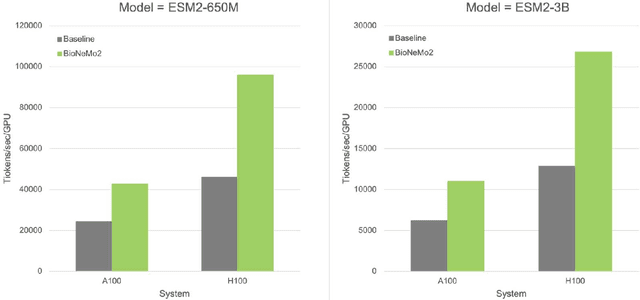
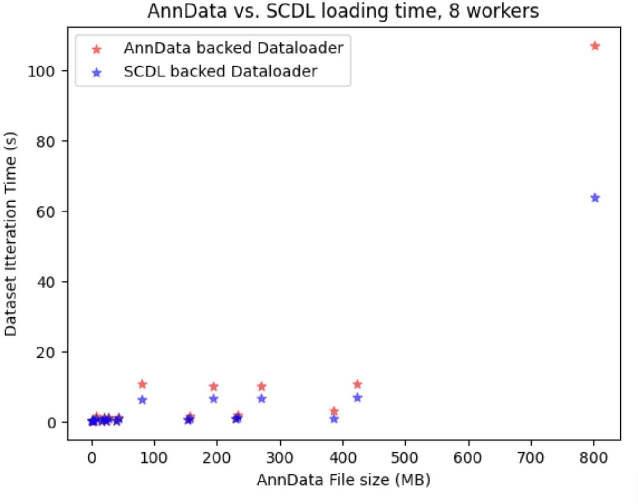
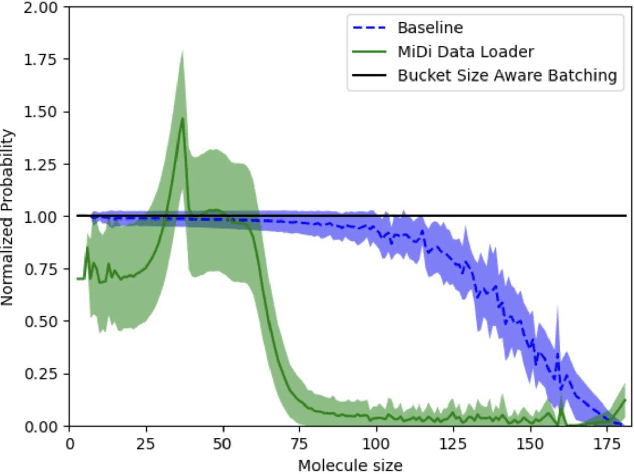
Artificial Intelligence models encoding biology and chemistry are opening new routes to high-throughput and high-quality in-silico drug development. However, their training increasingly relies on computational scale, with recent protein language models (pLM) training on hundreds of graphical processing units (GPUs). We introduce the BioNeMo Framework to facilitate the training of computational biology and chemistry AI models across hundreds of GPUs. Its modular design allows the integration of individual components, such as data loaders, into existing workflows and is open to community contributions. We detail technical features of the BioNeMo Framework through use cases such as pLM pre-training and fine-tuning. On 256 NVIDIA A100s, BioNeMo Framework trains a three billion parameter BERT-based pLM on over one trillion tokens in 4.2 days. The BioNeMo Framework is open-source and free for everyone to use.
EquiJump: Protein Dynamics Simulation via SO(3)-Equivariant Stochastic Interpolants
Oct 12, 2024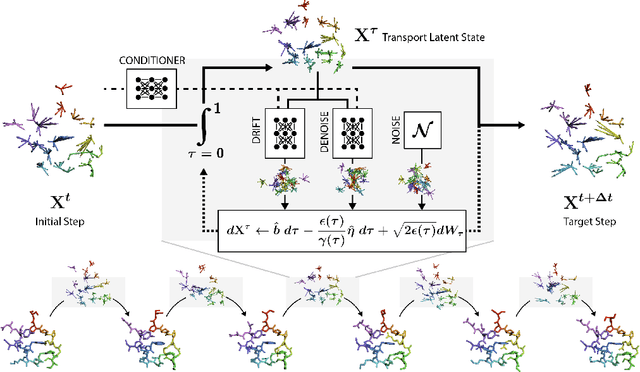

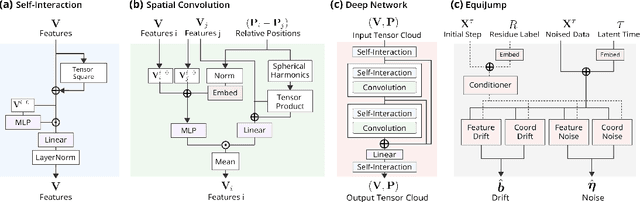
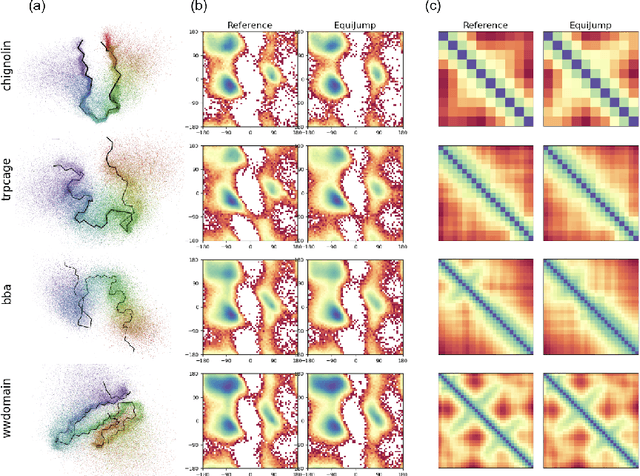
Mapping the conformational dynamics of proteins is crucial for elucidating their functional mechanisms. While Molecular Dynamics (MD) simulation enables detailed time evolution of protein motion, its computational toll hinders its use in practice. To address this challenge, multiple deep learning models for reproducing and accelerating MD have been proposed drawing on transport-based generative methods. However, existing work focuses on generation through transport of samples from prior distributions, that can often be distant from the data manifold. The recently proposed framework of stochastic interpolants, instead, enables transport between arbitrary distribution endpoints. Building upon this work, we introduce EquiJump, a transferable SO(3)-equivariant model that bridges all-atom protein dynamics simulation time steps directly. Our approach unifies diverse sampling methods and is benchmarked against existing models on trajectory data of fast folding proteins. EquiJump achieves state-of-the-art results on dynamics simulation with a transferable model on all of the fast folding proteins.
Large-scale Pretraining Improves Sample Efficiency of Active Learning based Molecule Virtual Screening
Sep 20, 2023Virtual screening of large compound libraries to identify potential hit candidates is one of the earliest steps in drug discovery. As the size of commercially available compound collections grows exponentially to the scale of billions, brute-force virtual screening using traditional tools such as docking becomes infeasible in terms of time and computational resources. Active learning and Bayesian optimization has recently been proven as effective methods of narrowing down the search space. An essential component in those methods is a surrogate machine learning model that is trained with a small subset of the library to predict the desired properties of compounds. Accurate model can achieve high sample efficiency by finding the most promising compounds with only a fraction of the whole library being virtually screened. In this study, we examined the performance of pretrained transformer-based language model and graph neural network in Bayesian optimization active learning framework. The best pretrained models identifies 58.97% of the top-50000 by docking score after screening only 0.6% of an ultra-large library containing 99.5 million compounds, improving 8% over previous state-of-the-art baseline. Through extensive benchmarks, we show that the superior performance of pretrained models persists in both structure-based and ligand-based drug discovery. Such model can serve as a boost to the accuracy and sample efficiency of active learning based molecule virtual screening.
Neural Network Predicts Ion Concentration Profiles under Nanoconfinement
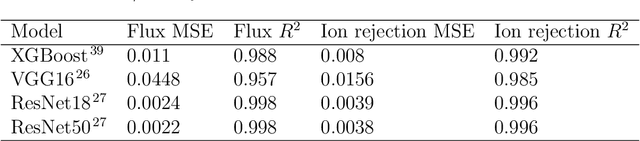
Apr 10, 2023Modeling the ion concentration profile in nanochannel plays an important role in understanding the electrical double layer and electroosmotic flow. Due to the non-negligible surface interaction and the effect of discrete solvent molecules, molecular dynamics (MD) simulation is often used as an essential tool to study the behavior of ions under nanoconfinement. Despite the accuracy of MD simulation in modeling nanoconfinement systems, it is computationally expensive. In this work, we propose neural network to predict ion concentration profiles in nanochannels with different configurations, including channel widths, ion molarity, and ion types. By modeling the ion concentration profile as a probability distribution, our neural network can serve as a much faster surrogate model for MD simulation with high accuracy. We further demonstrate the superior prediction accuracy of neural network over XGBoost. Lastly, we demonstrated that neural network is flexible in predicting ion concentration profiles with different bin sizes. Overall, our deep learning model is a fast, flexible, and accurate surrogate model to predict ion concentration profiles in nanoconfinement.
MOFormer: Self-Supervised Transformer model for Metal-Organic Framework Property Prediction
Oct 25, 2022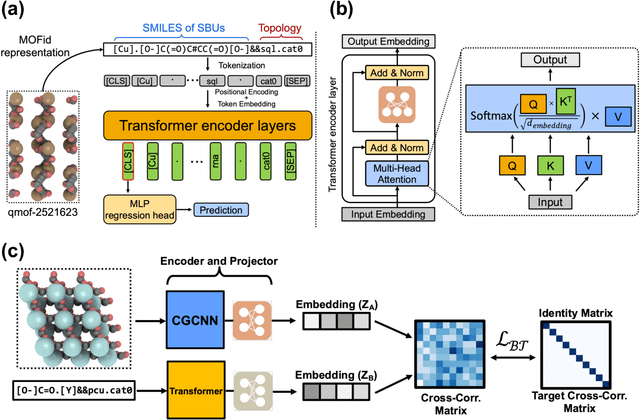


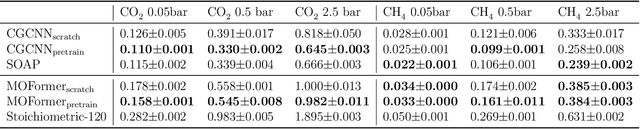
Metal-Organic Frameworks (MOFs) are materials with a high degree of porosity that can be used for applications in energy storage, water desalination, gas storage, and gas separation. However, the chemical space of MOFs is close to an infinite size due to the large variety of possible combinations of building blocks and topology. Discovering the optimal MOFs for specific applications requires an efficient and accurate search over an enormous number of potential candidates. Previous high-throughput screening methods using computational simulations like DFT can be time-consuming. Such methods also require optimizing 3D atomic structure of MOFs, which adds one extra step when evaluating hypothetical MOFs. In this work, we propose a structure-agnostic deep learning method based on the Transformer model, named as MOFormer, for property predictions of MOFs. The MOFormer takes a text string representation of MOF (MOFid) as input, thus circumventing the need of obtaining the 3D structure of hypothetical MOF and accelerating the screening process. Furthermore, we introduce a self-supervised learning framework that pretrains the MOFormer via maximizing the cross-correlation between its structure-agnostic representations and structure-based representations of crystal graph convolutional neural network (CGCNN) on >400k publicly available MOF data. Using self-supervised learning allows the MOFormer to intrinsically learn 3D structural information though it is not included in the input. Experiments show that pretraining improved the prediction accuracy of both models on various downstream prediction tasks. Furthermore, we revealed that MOFormer can be more data-efficient on quantum-chemical property prediction than structure-based CGCNN when training data is limited. Overall, MOFormer provides a novel perspective on efficient MOF design using deep learning.
MolCLR: Molecular Contrastive Learning of Representations via Graph Neural Networks
Feb 19, 2021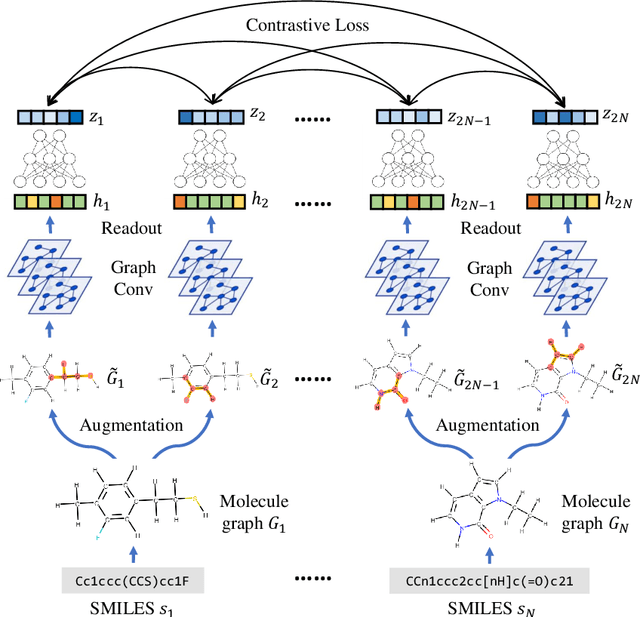
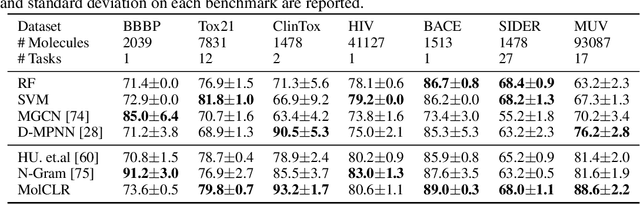
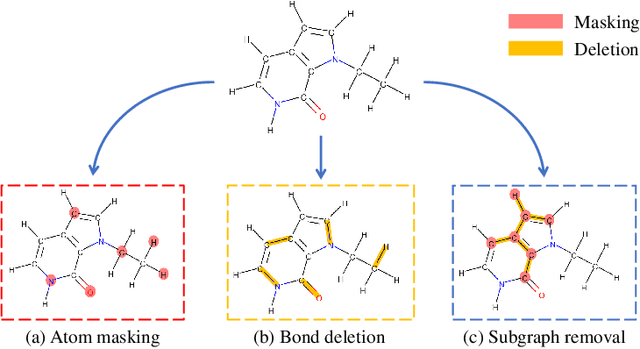

Molecular machine learning bears promise for efficient molecule property prediction and drug discovery. However, due to the limited labeled data and the giant chemical space, machine learning models trained via supervised learning perform poorly in generalization. This greatly limits the applications of machine learning methods for molecular design and discovery. In this work, we present MolCLR: Molecular Contrastive Learning of Representations via Graph Neural Networks (GNNs), a self-supervised learning framework for large unlabeled molecule datasets. Specifically, we first build a molecular graph, where each node represents an atom and each edge represents a chemical bond. A GNN is then used to encode the molecule graph. We propose three novel molecule graph augmentations: atom masking, bond deletion, and subgraph removal. A contrastive estimator is utilized to maximize the agreement of different graph augmentations from the same molecule. Experiments show that molecule representations learned by MolCLR can be transferred to multiple downstream molecular property prediction tasks. Our method thus achieves state-of-the-art performance on many challenging datasets. We also prove the efficiency of our proposed molecule graph augmentations on supervised molecular classification tasks.
Deep Reinforcement Learning Optimizes Graphene Nanopores for Efficient Desalination
Feb 09, 2021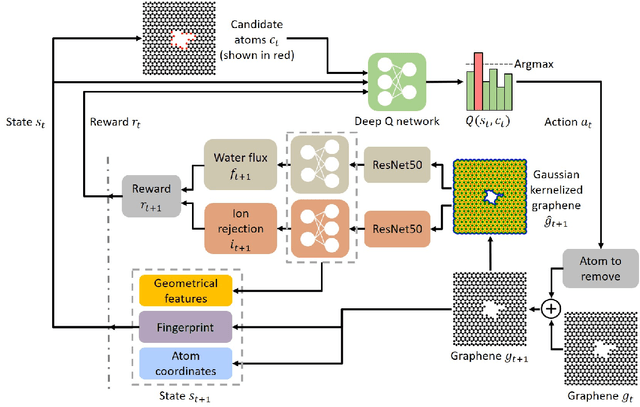
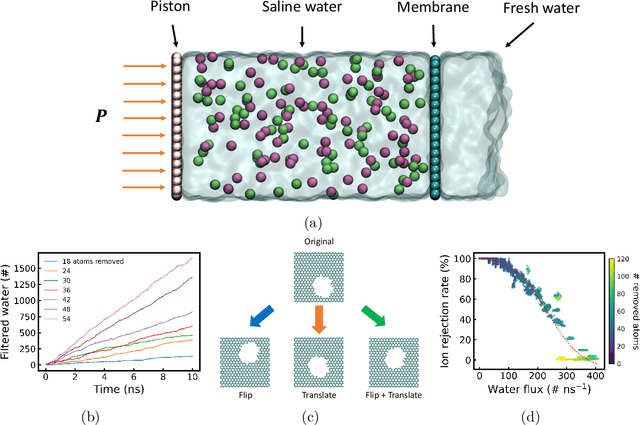
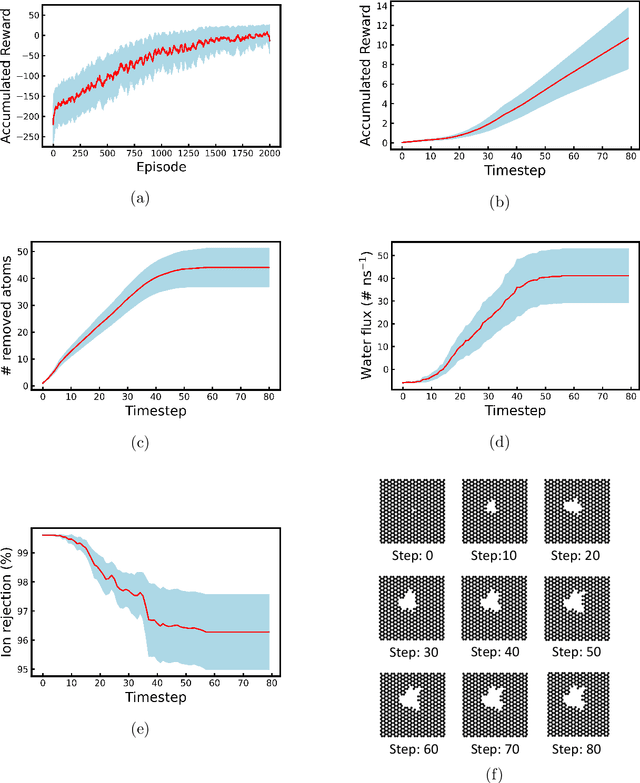
Two-dimensional nanomaterials, such as graphene, have been extensively studied because of their outstanding physical properties. Structure and geometry optimization of nanopores on such materials is beneficial for their performances in real-world engineering applications, like water desalination. However, the optimization process often involves very large number of experiments or simulations which are expensive and time-consuming. In this work, we propose a graphene nanopore optimization framework via the combination of deep reinforcement learning (DRL) and convolutional neural network (CNN) for efficient water desalination. The DRL agent controls the growth of nanopore by determining the atom to be removed at each timestep, while the CNN predicts the performance of nanoporus graphene for water desalination: the water flux and ion rejection at a certain external pressure. With the synchronous feedback from CNN-accelerated desalination performance prediction, our DRL agent can optimize the nanoporous graphene efficiently in an online manner. Molecular dynamics (MD) simulations on promising DRL-designed graphene nanopores show that they have higher water flux while maintaining rival ion rejection rate compared to the normal circular nanopores. Semi-oval shape with rough edges geometry of DRL-designed pores is found to be the key factor for their high water desalination performance. Ultimately, this study shows that DRL can be a powerful tool for material design.