 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMOFormer: Self-Supervised Transformer model for Metal-Organic Framework Property Prediction
Paper and Code
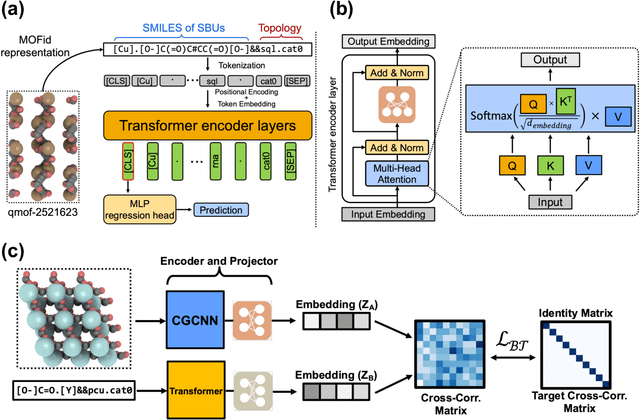


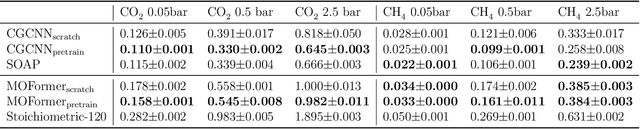
Metal-Organic Frameworks (MOFs) are materials with a high degree of porosity that can be used for applications in energy storage, water desalination, gas storage, and gas separation. However, the chemical space of MOFs is close to an infinite size due to the large variety of possible combinations of building blocks and topology. Discovering the optimal MOFs for specific applications requires an efficient and accurate search over an enormous number of potential candidates. Previous high-throughput screening methods using computational simulations like DFT can be time-consuming. Such methods also require optimizing 3D atomic structure of MOFs, which adds one extra step when evaluating hypothetical MOFs. In this work, we propose a structure-agnostic deep learning method based on the Transformer model, named as MOFormer, for property predictions of MOFs. The MOFormer takes a text string representation of MOF (MOFid) as input, thus circumventing the need of obtaining the 3D structure of hypothetical MOF and accelerating the screening process. Furthermore, we introduce a self-supervised learning framework that pretrains the MOFormer via maximizing the cross-correlation between its structure-agnostic representations and structure-based representations of crystal graph convolutional neural network (CGCNN) on >400k publicly available MOF data. Using self-supervised learning allows the MOFormer to intrinsically learn 3D structural information though it is not included in the input. Experiments show that pretraining improved the prediction accuracy of both models on various downstream prediction tasks. Furthermore, we revealed that MOFormer can be more data-efficient on quantum-chemical property prediction than structure-based CGCNN when training data is limited. Overall, MOFormer provides a novel perspective on efficient MOF design using deep learning.