 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFrom Knowledge to Action: Outcomes of the 2025 Large Language Model (LLM) Hackathon for Applications in Materials Science and Chemistry
May 04, 2026Large language models (LLMs) are rapidly changing how researchers in materials science and chemistry discover, organize, and act on scientific knowledge. This paper analyzes a broad set of community-developed LLM applications in an effort to identify emerging patterns in how these systems can be used across the scientific research lifecycle. We organize the projects into two complementary categories: Knowledge Infrastructure, systems that structure, retrieve, synthesize, and validate scientific information; and Action Systems, systems that execute, coordinate, or automate scientific work across computational and experimental environments. The submissions reveal a shift from single-purpose LLM tools toward integrated, multi-agent workflows that combine retrieval, reasoning, tool use, and domain-specific validation. Prominent themes include retrieval-augmented generation as grounding infrastructure, persistent structured knowledge representations, multimodal and multilingual scientific inputs, and early progress toward laboratory-integrated closed-loop systems. Together, these results suggest that LLMs are evolving from general-purpose assistants into composable infrastructure for scientific reasoning and action. This work provides a community snapshot of that transition and a practical taxonomy for understanding emerging LLM-enabled workflows in materials science and chemistry.
GPCR-BERT: Interpreting Sequential Design of G Protein Coupled Receptors Using Protein Language Models
Oct 30, 2023With the rise of Transformers and Large Language Models (LLMs) in Chemistry and Biology, new avenues for the design and understanding of therapeutics have opened up to the scientific community. Protein sequences can be modeled as language and can take advantage of recent advances in LLMs, specifically with the abundance of our access to the protein sequence datasets. In this paper, we developed the GPCR-BERT model for understanding the sequential design of G Protein-Coupled Receptors (GPCRs). GPCRs are the target of over one-third of FDA-approved pharmaceuticals. However, there is a lack of comprehensive understanding regarding the relationship between amino acid sequence, ligand selectivity, and conformational motifs (such as NPxxY, CWxP, E/DRY). By utilizing the pre-trained protein model (Prot-Bert) and fine-tuning with prediction tasks of variations in the motifs, we were able to shed light on several relationships between residues in the binding pocket and some of the conserved motifs. To achieve this, we took advantage of attention weights, and hidden states of the model that are interpreted to extract the extent of contributions of amino acids in dictating the type of masked ones. The fine-tuned models demonstrated high accuracy in predicting hidden residues within the motifs. In addition, the analysis of embedding was performed over 3D structures to elucidate the higher-order interactions within the conformations of the receptors.
Materials Informatics Transformer: A Language Model for Interpretable Materials Properties Prediction
Sep 01, 2023



Recently, the remarkable capabilities of large language models (LLMs) have been illustrated across a variety of research domains such as natural language processing, computer vision, and molecular modeling. We extend this paradigm by utilizing LLMs for material property prediction by introducing our model Materials Informatics Transformer (MatInFormer). Specifically, we introduce a novel approach that involves learning the grammar of crystallography through the tokenization of pertinent space group information. We further illustrate the adaptability of MatInFormer by incorporating task-specific data pertaining to Metal-Organic Frameworks (MOFs). Through attention visualization, we uncover the key features that the model prioritizes during property prediction. The effectiveness of our proposed model is empirically validated across 14 distinct datasets, hereby underscoring its potential for high throughput screening through accurate material property prediction.
MOFormer: Self-Supervised Transformer model for Metal-Organic Framework Property Prediction
Oct 25, 2022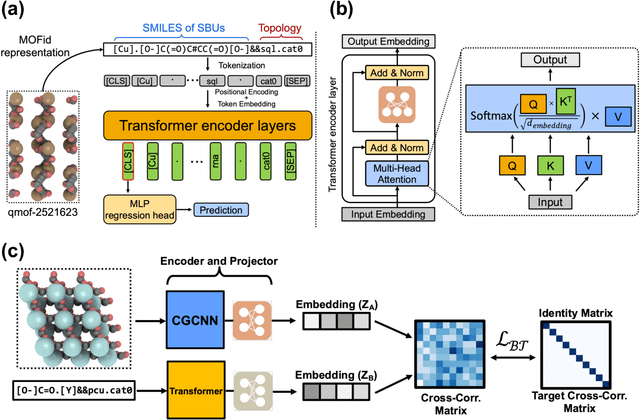


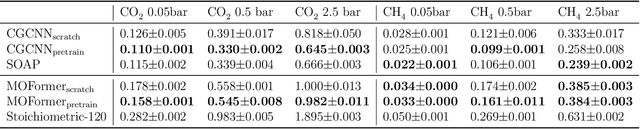
Metal-Organic Frameworks (MOFs) are materials with a high degree of porosity that can be used for applications in energy storage, water desalination, gas storage, and gas separation. However, the chemical space of MOFs is close to an infinite size due to the large variety of possible combinations of building blocks and topology. Discovering the optimal MOFs for specific applications requires an efficient and accurate search over an enormous number of potential candidates. Previous high-throughput screening methods using computational simulations like DFT can be time-consuming. Such methods also require optimizing 3D atomic structure of MOFs, which adds one extra step when evaluating hypothetical MOFs. In this work, we propose a structure-agnostic deep learning method based on the Transformer model, named as MOFormer, for property predictions of MOFs. The MOFormer takes a text string representation of MOF (MOFid) as input, thus circumventing the need of obtaining the 3D structure of hypothetical MOF and accelerating the screening process. Furthermore, we introduce a self-supervised learning framework that pretrains the MOFormer via maximizing the cross-correlation between its structure-agnostic representations and structure-based representations of crystal graph convolutional neural network (CGCNN) on >400k publicly available MOF data. Using self-supervised learning allows the MOFormer to intrinsically learn 3D structural information though it is not included in the input. Experiments show that pretraining improved the prediction accuracy of both models on various downstream prediction tasks. Furthermore, we revealed that MOFormer can be more data-efficient on quantum-chemical property prediction than structure-based CGCNN when training data is limited. Overall, MOFormer provides a novel perspective on efficient MOF design using deep learning.
Crystal Twins: Self-supervised Learning for Crystalline Material Property Prediction
May 04, 2022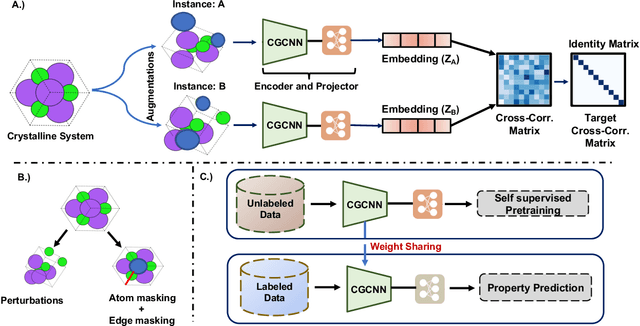
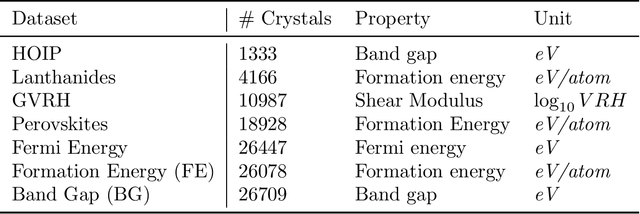
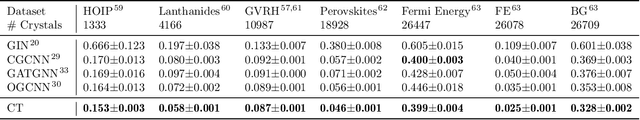
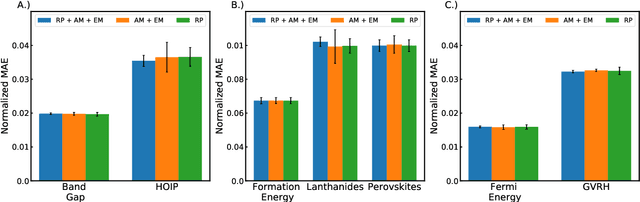
Machine learning (ML) models have been widely successful in the prediction of material properties. However, large labeled datasets required for training accurate ML models are elusive and computationally expensive to generate. Recent advances in Self-Supervised Learning (SSL) frameworks capable of training ML models on unlabeled data have mitigated this problem and demonstrated superior performance in computer vision and natural language processing tasks. Drawing inspiration from the developments in SSL, we introduce Crystal Twins (CT): an SSL method for crystalline materials property prediction. Using a large unlabeled dataset, we pre-train a Graph Neural Network (GNN) by applying the redundancy reduction principle to the graph latent embeddings of augmented instances obtained from the same crystalline system. By sharing the pre-trained weights when fine-tuning the GNN for regression tasks, we significantly improve the performance for 7 challenging material property prediction benchmarks
Improving Molecular Contrastive Learning via Faulty Negative Mitigation and Decomposed Fragment Contrast
Feb 18, 2022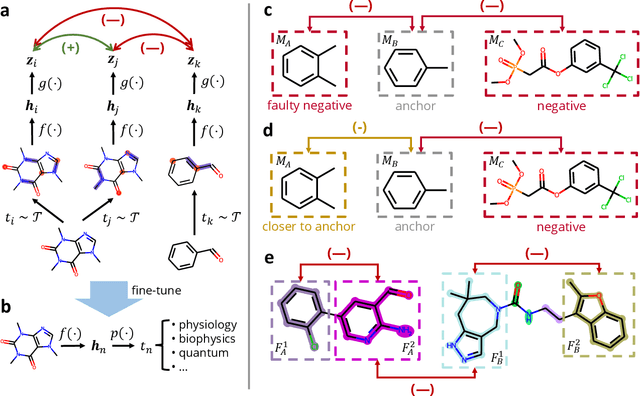
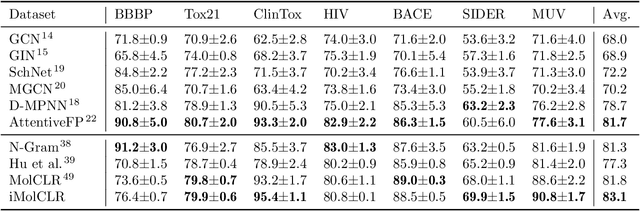
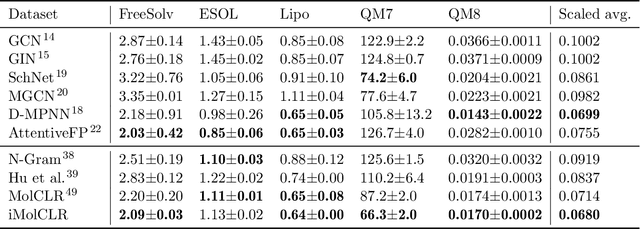
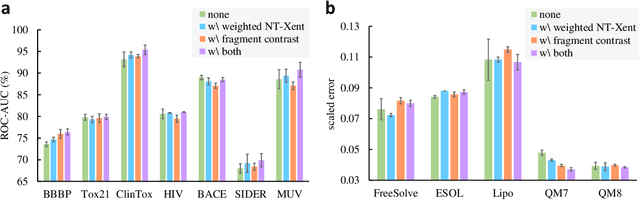
Deep learning has been a prevalence in computational chemistry and widely implemented in molecule property predictions. Recently, self-supervised learning (SSL), especially contrastive learning (CL), gathers growing attention for the potential to learn molecular representations that generalize to the gigantic chemical space. Unlike supervised learning, SSL can directly leverage large unlabeled data, which greatly reduces the effort to acquire molecular property labels through costly and time-consuming simulations or experiments. However, most molecular SSL methods borrow the insights from the machine learning community but neglect the unique cheminformatics (e.g., molecular fingerprints) and multi-level graphical structures (e.g., functional groups) of molecules. In this work, we propose iMolCLR: improvement of Molecular Contrastive Learning of Representations with graph neural networks (GNNs) in two aspects, (1) mitigating faulty negative contrastive instances via considering cheminformatics similarities between molecule pairs; (2) fragment-level contrasting between intra- and inter-molecule substructures decomposed from molecules. Experiments have shown that the proposed strategies significantly improve the performance of GNN models on various challenging molecular property predictions. In comparison to the previous CL framework, iMolCLR demonstrates an averaged 1.3% improvement of ROC-AUC on 7 classification benchmarks and an averaged 4.8% decrease of the error on 5 regression benchmarks. On most benchmarks, the generic GNN pre-trained by iMolCLR rivals or even surpasses supervised learning models with sophisticated architecture designs and engineered features. Further investigations demonstrate that representations learned through iMolCLR intrinsically embed scaffolds and functional groups that can reason molecule similarities.
AugLiChem: Data Augmentation Library of Chemical Structures for Machine Learning
Dec 01, 2021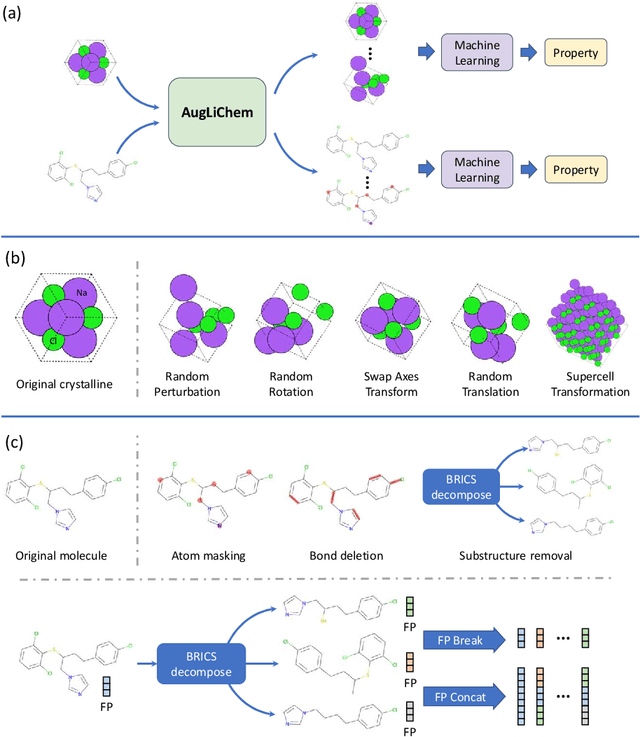
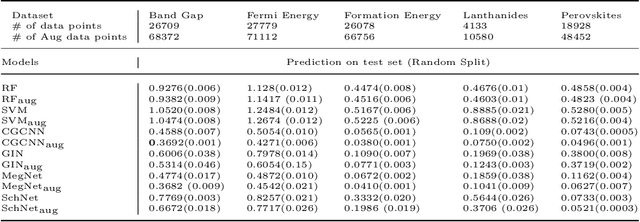
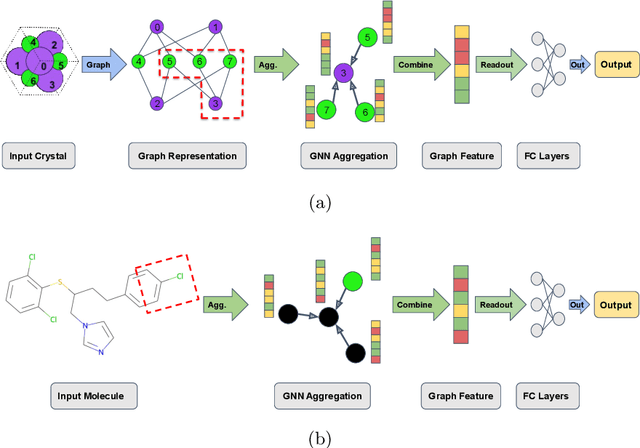
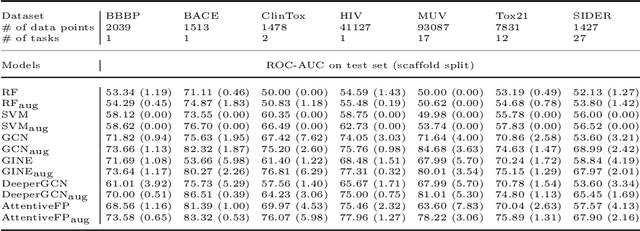
Machine learning (ML) has demonstrated the promise for accurate and efficient property prediction of molecules and crystalline materials. To develop highly accurate ML models for chemical structure property prediction, datasets with sufficient samples are required. However, obtaining clean and sufficient data of chemical properties can be expensive and time-consuming, which greatly limits the performance of ML models. Inspired by the success of data augmentations in computer vision and natural language processing, we developed AugLiChem: the data augmentation library for chemical structures. Augmentation methods for both crystalline systems and molecules are introduced, which can be utilized for fingerprint-based ML models and Graph Neural Networks(GNNs). We show that using our augmentation strategies significantly improves the performance of ML models, especially when using GNNs. In addition, the augmentations that we developed can be used as a direct plug-in module during training and have demonstrated the effectiveness when implemented with different GNN models through the AugliChem library. The Python-based package for our implementation of Auglichem: Data augmentation library for chemical structures, is publicly available at: https://github.com/BaratiLab/AugLiChem.
FaultNet: A Deep Convolutional Neural Network for bearing fault classification
Oct 05, 2020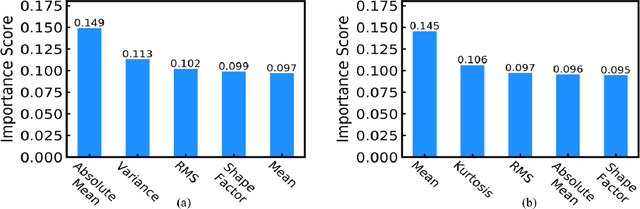
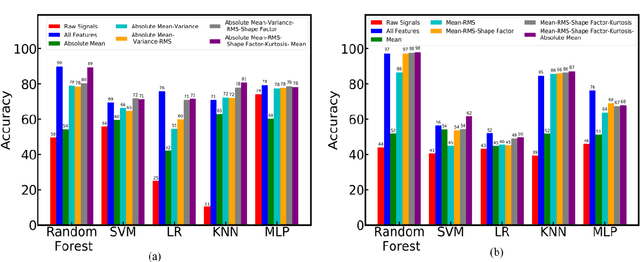
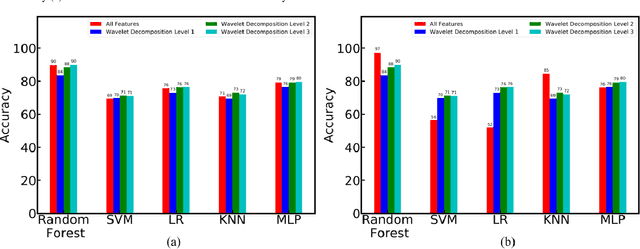
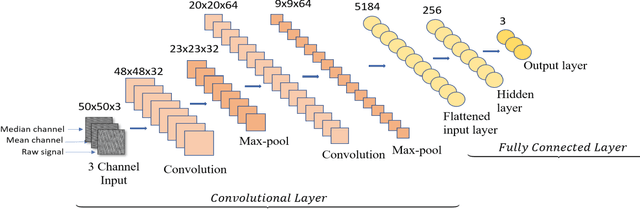
The increased presence of advanced sensors on the production floors has led to collection of datasets that can provide significant insights into machine health. An important and reliable indicator of machine health, vibration signal data can provide us a greater understanding of different faults occurring in mechanical systems. In this work, we analyze vibration signal data of mechanical systems with bearings by combining different signal processing techniques and coupling them machine learning techniques to classify different types of bearing faults. We also highlight the importance of using of different signal processing methods and analyze their effect on bearing fault detection. Apart from the traditional machine learning algorithms we also propose a convolutional neural network FaultNet which can effectively determine the type of bearing fault with a high degree of accuracy. The distinguishing factor of this work is the idea of channels proposed to extract more information from the signal, we have stacked the mean and median channels to raw signal to extract more useful features to classify the signals with greater accuracy
W-Net: Dense Semantic Segmentation of Subcutaneous Tissue in Ultrasound Images by Expanding U-Net to Incorporate Ultrasound RF Waveform Data
Sep 02, 2020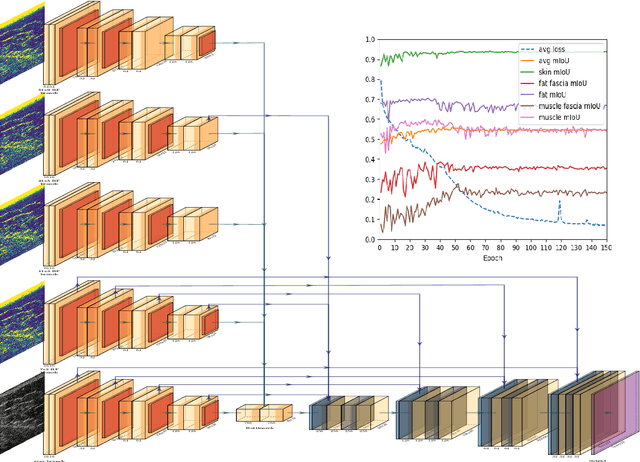
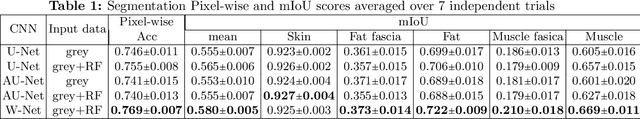
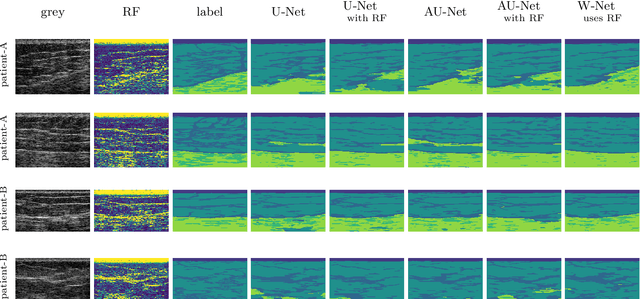
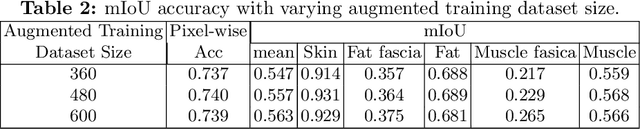
We present W-Net, a novel Convolution Neural Network (CNN) framework that employs raw ultrasound waveforms from each A-scan, typically referred to as ultrasound Radio Frequency (RF) data, in addition to the gray ultrasound image to semantically segment and label tissues. Unlike prior work, we seek to label every pixel in the image, without the use of a background class. To the best of our knowledge, this is also the first deep-learning or CNN approach for segmentation that analyses ultrasound raw RF data along with the gray image. International patent(s) pending [PCT/US20/37519]. We chose subcutaneous tissue (SubQ) segmentation as our initial clinical goal since it has diverse intermixed tissues, is challenging to segment, and is an underrepresented research area. SubQ potential applications include plastic surgery, adipose stem-cell harvesting, lymphatic monitoring, and possibly detection/treatment of certain types of tumors. A custom dataset consisting of hand-labeled images by an expert clinician and trainees are used for the experimentation, currently labeled into the following categories: skin, fat, fat fascia/stroma, muscle and muscle fascia. We compared our results with U-Net and Attention U-Net. Our novel \emph{W-Net}'s RF-Waveform input and architecture increased mIoU accuracy (averaged across all tissue classes) by 4.5\% and 4.9\% compared to regular U-Net and Attention U-Net, respectively. We present analysis as to why the Muscle fascia and Fat fascia/stroma are the most difficult tissues to label. Muscle fascia in particular, the most difficult anatomic class to recognize for both humans and AI algorithms, saw mIoU improvements of 13\% and 16\% from our W-Net vs U-Net and Attention U-Net respectively.
Potential Neutralizing Antibodies Discovered for Novel Corona Virus Using Machine Learning
Mar 18, 2020

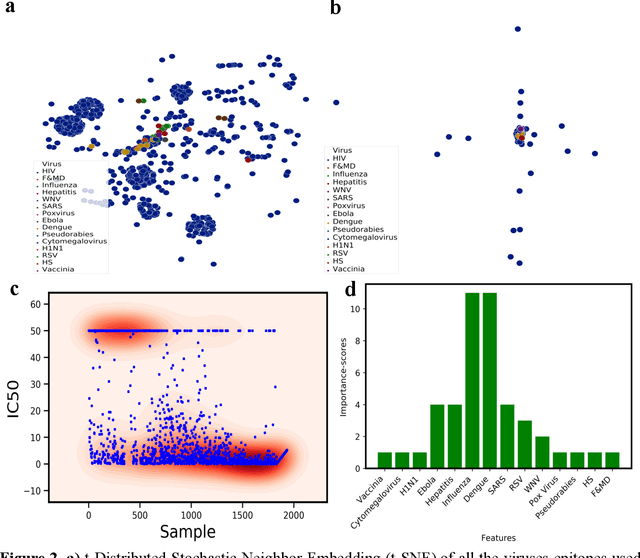
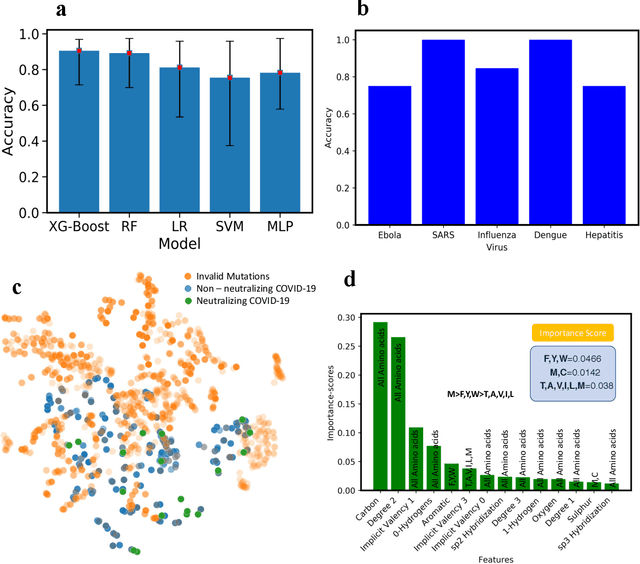
The fast and untraceable virus mutations take lives of thousands of people before the immune system can produce the inhibitory antibody. Recent outbreak of novel coronavirus infected and killed thousands of people in the world. Rapid methods in finding peptides or antibody sequences that can inhibit the viral epitopes of COVID-19 will save the life of thousands. In this paper, we devised a machine learning (ML) model to predict the possible inhibitory synthetic antibodies for Corona virus. We collected 1933 virus-antibody sequences and their clinical patient neutralization response and trained an ML model to predict the antibody response. Using graph featurization with variety of ML methods, we screened thousands of hypothetical antibody sequences and found 8 stable antibodies that potentially inhibit COVID-19. We combined bioinformatics, structural biology, and Molecular Dynamics (MD) simulations to verify the stability of the candidate antibodies that can inhibit the Corona virus.