 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMaterials Informatics Transformer: A Language Model for Interpretable Materials Properties Prediction
Sep 01, 2023



Recently, the remarkable capabilities of large language models (LLMs) have been illustrated across a variety of research domains such as natural language processing, computer vision, and molecular modeling. We extend this paradigm by utilizing LLMs for material property prediction by introducing our model Materials Informatics Transformer (MatInFormer). Specifically, we introduce a novel approach that involves learning the grammar of crystallography through the tokenization of pertinent space group information. We further illustrate the adaptability of MatInFormer by incorporating task-specific data pertaining to Metal-Organic Frameworks (MOFs). Through attention visualization, we uncover the key features that the model prioritizes during property prediction. The effectiveness of our proposed model is empirically validated across 14 distinct datasets, hereby underscoring its potential for high throughput screening through accurate material property prediction.
Denoise Pre-training on Non-equilibrium Molecules for Accurate and Transferable Neural Potentials
Mar 03, 2023Machine learning methods, particularly recent advances in equivariant graph neural networks (GNNs), have been investigated as surrogate models to expensive ab initio quantum mechanics (QM) approaches for molecular potential predictions. However, building accurate and transferable potential models using GNNs remains challenging, as the quality and quantity of data are greatly limited by QM calculations, especially for large and complex molecular systems. In this work, we propose denoise pre-training on non-equilibrium molecular conformations to achieve more accurate and transferable GNN potential predictions. Specifically, GNNs are pre-trained by predicting the random noises added to atomic coordinates of sampled non-equilibrium conformations. Rigorous experiments on multiple benchmarks reveal that pre-training significantly improves the accuracy of neural potentials. Furthermore, we show that the proposed pre-training approach is model-agnostic, as it improves the performance of different invariant and equivariant GNNs. Notably, our models pre-trained on small molecules demonstrate remarkable transferability, improving performance when fine-tuned on diverse molecular systems, including different elements, charged molecules, biomolecules, and larger systems. These results highlight the potential for leveraging denoise pre-training approaches to build more generalizable neural potentials for complex molecular systems.
TransPolymer: a Transformer-based Language Model for Polymer Property Predictions
Sep 11, 2022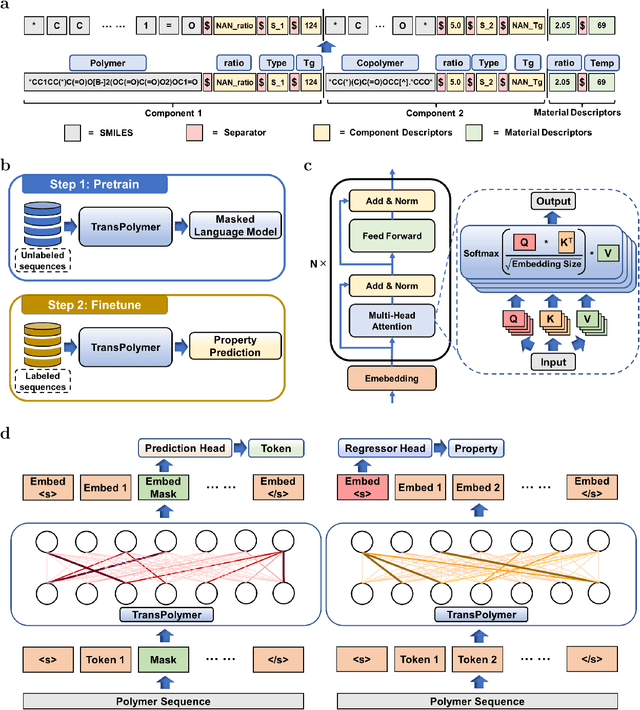
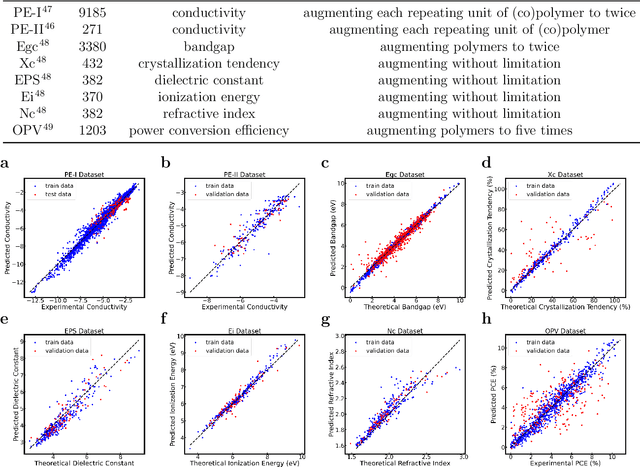
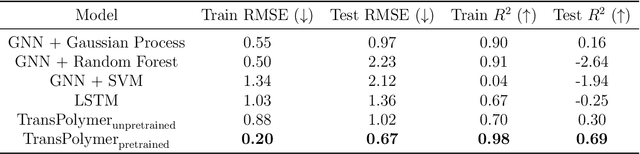
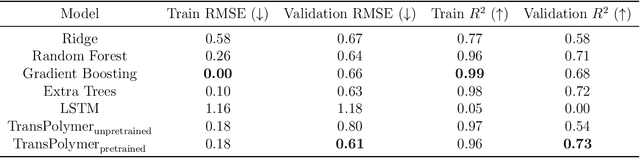
Accurate and efficient prediction of polymer properties is of great significance in polymer development and design. Conventionally, expensive and time-consuming experiments or simulations are required to evaluate the function of polymers. Recently, Transformer models, equipped with attention mechanisms, have exhibited superior performance in various natural language processing tasks. However, such methods have not been investigated in polymer sciences. Herein, we report TransPolymer, a Transformer-based language model for polymer property prediction. Owing to our proposed polymer tokenizer with chemical awareness, TransPolymer can learn representations directly from polymer sequences. The model learns expressive representations by pretraining on a large unlabeled dataset, followed by finetuning the model on downstream datasets concerning various polymer properties. TransPolymer achieves superior performance in all eight datasets and surpasses other baselines significantly on most downstream tasks. Moreover, the improvement by the pretrained TransPolymer over supervised TransPolymer and other language models strengthens the significant benefits of pretraining on large unlabeled data in representation learning. Experiment results further demonstrate the important role of the attention mechanism in understanding polymer sequences. We highlight this model as a promising computational tool for promoting rational polymer design and understanding structure-property relationships in a data science view.