 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEquiJump: Protein Dynamics Simulation via SO(3)-Equivariant Stochastic Interpolants
Oct 12, 2024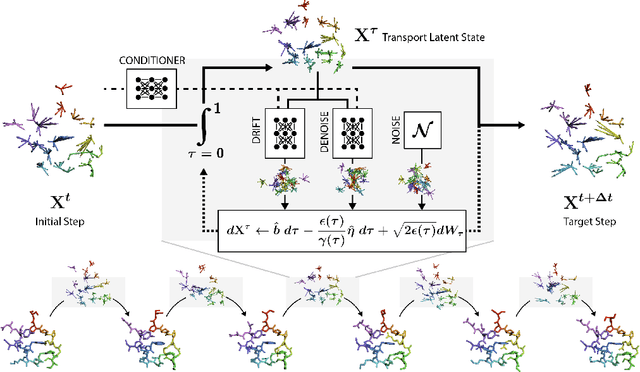

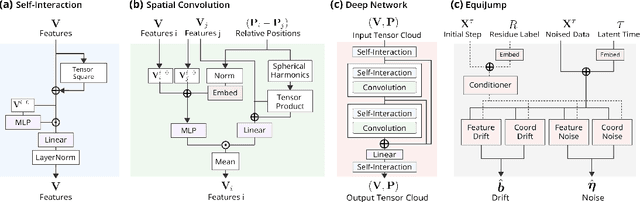
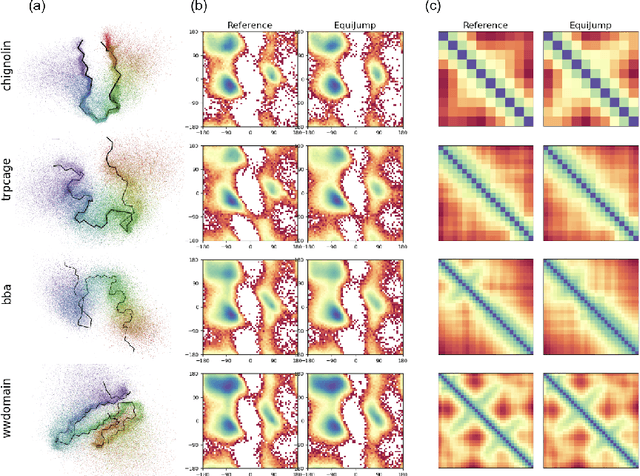
Mapping the conformational dynamics of proteins is crucial for elucidating their functional mechanisms. While Molecular Dynamics (MD) simulation enables detailed time evolution of protein motion, its computational toll hinders its use in practice. To address this challenge, multiple deep learning models for reproducing and accelerating MD have been proposed drawing on transport-based generative methods. However, existing work focuses on generation through transport of samples from prior distributions, that can often be distant from the data manifold. The recently proposed framework of stochastic interpolants, instead, enables transport between arbitrary distribution endpoints. Building upon this work, we introduce EquiJump, a transferable SO(3)-equivariant model that bridges all-atom protein dynamics simulation time steps directly. Our approach unifies diverse sampling methods and is benchmarked against existing models on trajectory data of fast folding proteins. EquiJump achieves state-of-the-art results on dynamics simulation with a transferable model on all of the fast folding proteins.
E3STO: Orbital Inspired SE(3)-Equivariant Molecular Representation for Electron Density Prediction
Oct 08, 2024



Electron density prediction stands as a cornerstone challenge in molecular systems, pivotal for various applications such as understanding molecular interactions and conducting precise quantum mechanical calculations. However, the scaling of density functional theory (DFT) calculations is prohibitively expensive. Machine learning methods provide an alternative, offering efficiency and accuracy. We introduce a novel SE(3)-equivariant architecture, drawing inspiration from Slater-Type Orbitals (STO), to learn representations of molecular electronic structures. Our approach offers an alternative functional form for learned orbital-like molecular representation. We showcase the effectiveness of our method by achieving SOTA prediction accuracy of molecular electron density with 30-70\% improvement over other work on Molecular Dynamics data.
Ophiuchus: Scalable Modeling of Protein Structures through Hierarchical Coarse-graining SO(3)-Equivariant Autoencoders
Oct 04, 2023Three-dimensional native states of natural proteins display recurring and hierarchical patterns. Yet, traditional graph-based modeling of protein structures is often limited to operate within a single fine-grained resolution, and lacks hourglass neural architectures to learn those high-level building blocks. We narrow this gap by introducing Ophiuchus, an SO(3)-equivariant coarse-graining model that efficiently operates on all heavy atoms of standard protein residues, while respecting their relevant symmetries. Our model departs from current approaches that employ graph modeling, instead focusing on local convolutional coarsening to model sequence-motif interactions in log-linear length complexity. We train Ophiuchus on contiguous fragments of PDB monomers, investigating its reconstruction capabilities across different compression rates. We examine the learned latent space and demonstrate its prompt usage in conformational interpolation, comparing interpolated trajectories to structure snapshots from the PDBFlex dataset. Finally, we leverage denoising diffusion probabilistic models (DDPM) to efficiently sample readily-decodable latent embeddings of diverse miniproteins. Our experiments demonstrate Ophiuchus to be a scalable basis for efficient protein modeling and generation.