 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAccelerating Protein Molecular Dynamics Simulation with DeepJump
Sep 16, 2025


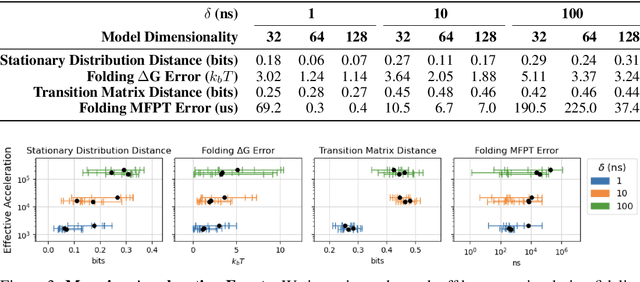
Unraveling the dynamical motions of biomolecules is essential for bridging their structure and function, yet it remains a major computational challenge. Molecular dynamics (MD) simulation provides a detailed depiction of biomolecular motion, but its high-resolution temporal evolution comes at significant computational cost, limiting its applicability to timescales of biological relevance. Deep learning approaches have emerged as promising solutions to overcome these computational limitations by learning to predict long-timescale dynamics. However, generalizable kinetics models for proteins remain largely unexplored, and the fundamental limits of achievable acceleration while preserving dynamical accuracy are poorly understood. In this work, we fill this gap with DeepJump, an Euclidean-Equivariant Flow Matching-based model for predicting protein conformational dynamics across multiple temporal scales. We train DeepJump on trajectories of the diverse proteins of mdCATH, systematically studying our model's performance in generalizing to long-term dynamics of fast-folding proteins and characterizing the trade-off between computational acceleration and prediction accuracy. We demonstrate the application of DeepJump to ab initio folding, showcasing prediction of folding pathways and native states. Our results demonstrate that DeepJump achieves significant $\approx$1000$\times$ computational acceleration while effectively recovering long-timescale dynamics, providing a stepping stone for enabling routine simulation of proteins.
RiboGen: RNA Sequence and Structure Co-Generation with Equivariant MultiFlow
Mar 03, 2025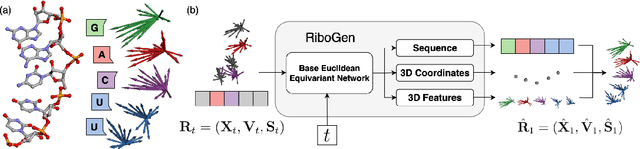
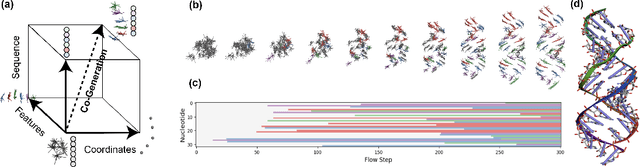
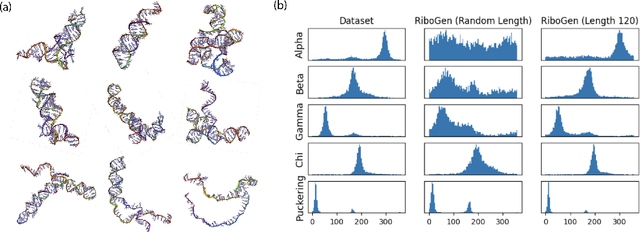
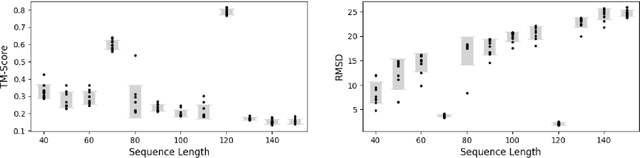
Ribonucleic acid (RNA) plays fundamental roles in biological systems, from carrying genetic information to performing enzymatic function. Understanding and designing RNA can enable novel therapeutic application and biotechnological innovation. To enhance RNA design, in this paper we introduce RiboGen, the first deep learning model to simultaneously generate RNA sequence and all-atom 3D structure. RiboGen leverages the standard Flow Matching with Discrete Flow Matching in a multimodal data representation. RiboGen is based on Euclidean Equivariant neural networks for efficiently processing and learning three-dimensional geometry. Our experiments show that RiboGen can efficiently generate chemically plausible and self-consistent RNA samples. Our results suggest that co-generation of sequence and structure is a competitive approach for modeling RNA.
BioNeMo Framework: a modular, high-performance library for AI model development in drug discovery
Nov 15, 2024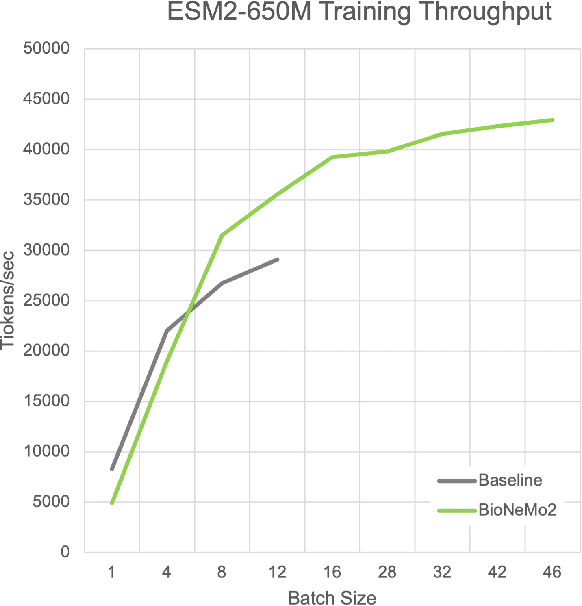
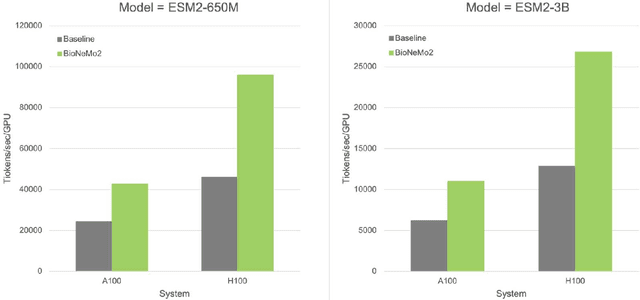
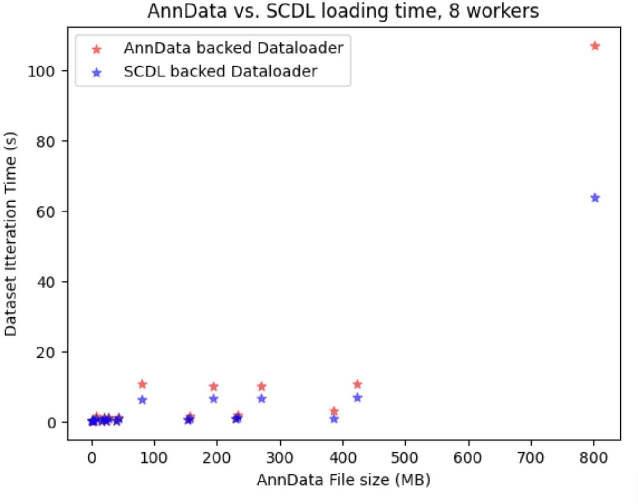
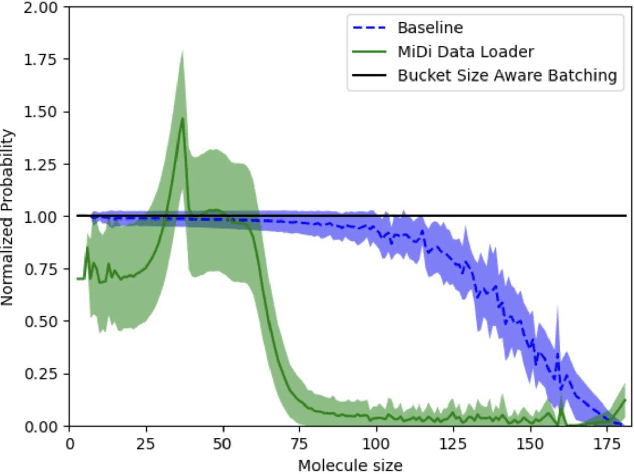
Artificial Intelligence models encoding biology and chemistry are opening new routes to high-throughput and high-quality in-silico drug development. However, their training increasingly relies on computational scale, with recent protein language models (pLM) training on hundreds of graphical processing units (GPUs). We introduce the BioNeMo Framework to facilitate the training of computational biology and chemistry AI models across hundreds of GPUs. Its modular design allows the integration of individual components, such as data loaders, into existing workflows and is open to community contributions. We detail technical features of the BioNeMo Framework through use cases such as pLM pre-training and fine-tuning. On 256 NVIDIA A100s, BioNeMo Framework trains a three billion parameter BERT-based pLM on over one trillion tokens in 4.2 days. The BioNeMo Framework is open-source and free for everyone to use.
EquiJump: Protein Dynamics Simulation via SO(3)-Equivariant Stochastic Interpolants
Oct 12, 2024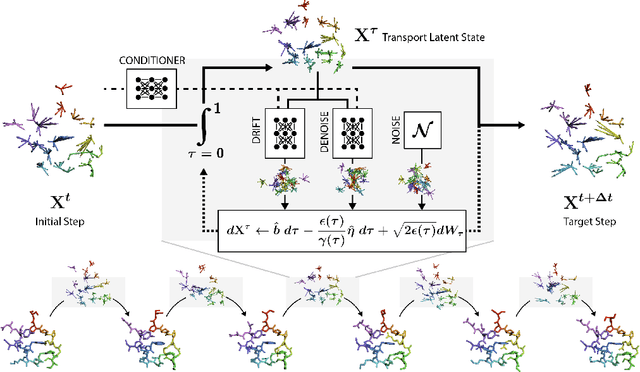

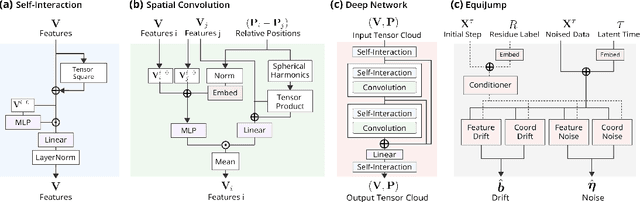
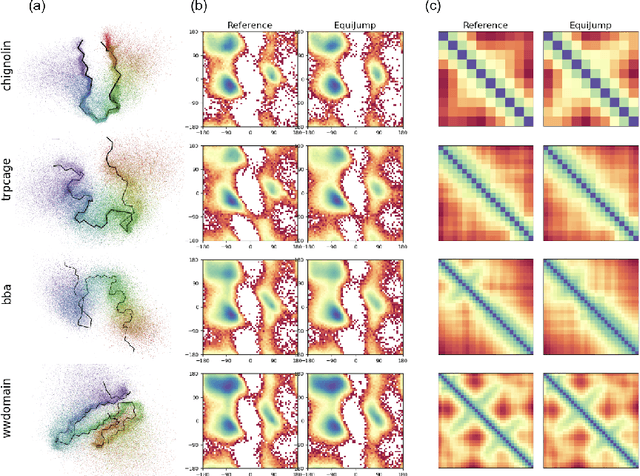
Mapping the conformational dynamics of proteins is crucial for elucidating their functional mechanisms. While Molecular Dynamics (MD) simulation enables detailed time evolution of protein motion, its computational toll hinders its use in practice. To address this challenge, multiple deep learning models for reproducing and accelerating MD have been proposed drawing on transport-based generative methods. However, existing work focuses on generation through transport of samples from prior distributions, that can often be distant from the data manifold. The recently proposed framework of stochastic interpolants, instead, enables transport between arbitrary distribution endpoints. Building upon this work, we introduce EquiJump, a transferable SO(3)-equivariant model that bridges all-atom protein dynamics simulation time steps directly. Our approach unifies diverse sampling methods and is benchmarked against existing models on trajectory data of fast folding proteins. EquiJump achieves state-of-the-art results on dynamics simulation with a transferable model on all of the fast folding proteins.
Ophiuchus: Scalable Modeling of Protein Structures through Hierarchical Coarse-graining SO(3)-Equivariant Autoencoders
Oct 04, 2023Three-dimensional native states of natural proteins display recurring and hierarchical patterns. Yet, traditional graph-based modeling of protein structures is often limited to operate within a single fine-grained resolution, and lacks hourglass neural architectures to learn those high-level building blocks. We narrow this gap by introducing Ophiuchus, an SO(3)-equivariant coarse-graining model that efficiently operates on all heavy atoms of standard protein residues, while respecting their relevant symmetries. Our model departs from current approaches that employ graph modeling, instead focusing on local convolutional coarsening to model sequence-motif interactions in log-linear length complexity. We train Ophiuchus on contiguous fragments of PDB monomers, investigating its reconstruction capabilities across different compression rates. We examine the learned latent space and demonstrate its prompt usage in conformational interpolation, comparing interpolated trajectories to structure snapshots from the PDBFlex dataset. Finally, we leverage denoising diffusion probabilistic models (DDPM) to efficiently sample readily-decodable latent embeddings of diverse miniproteins. Our experiments demonstrate Ophiuchus to be a scalable basis for efficient protein modeling and generation.