 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAdvancing Ligand-based Virtual Screening and Molecular Generation with Pretrained Molecular Embedding Distance
Apr 27, 2026Molecular similarity plays a central role in ligand-based drug discovery, such as virtual screening, analog searching, and goal-directed molecular generation. However, traditional similarity measures, ranging from fingerprint-based Tanimoto coefficients to 3D shape overlays, are often computationally expensive at scale or rely on hand-crafted molecular descriptors. Meanwhile, many deep learning approaches to similarity-aware design still depend on similarity-specific supervision or costly data curation, limiting their generality across targets. In this work, we propose pretrained embedding distance (PED) as an effective alternative, computed directly from pretrained molecular models without task-specific training. Experimental results show that PED exhibits distinct correlations with traditional similarity metrics, and performs effectively in both ranking molecules for virtual screening and guiding molecular generation via reward design. These findings suggest that pretrained molecular embeddings capture rich structural information and can serve as a promising and scalable similarity measurement for modern AI-aided drug discovery.
ACEGEN: Reinforcement learning of generative chemical agents for drug discovery
May 07, 2024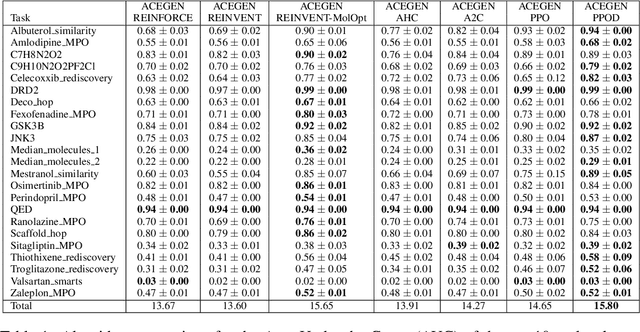


In recent years, reinforcement learning (RL) has emerged as a valuable tool in drug design, offering the potential to propose and optimize molecules with desired properties. However, striking a balance between capability, flexibility, and reliability remains challenging due to the complexity of advanced RL algorithms and the significant reliance on specialized code. In this work, we introduce ACEGEN, a comprehensive and streamlined toolkit tailored for generative drug design, built using TorchRL, a modern decision-making library that offers efficient and thoroughly tested reusable components. ACEGEN provides a robust, flexible, and efficient platform for molecular design. We validate its effectiveness by benchmarking it across various algorithms and conducting multiple drug discovery case studies. ACEGEN is accessible at https://github.com/acellera/acegen-open.
Large-scale Pretraining Improves Sample Efficiency of Active Learning based Molecule Virtual Screening
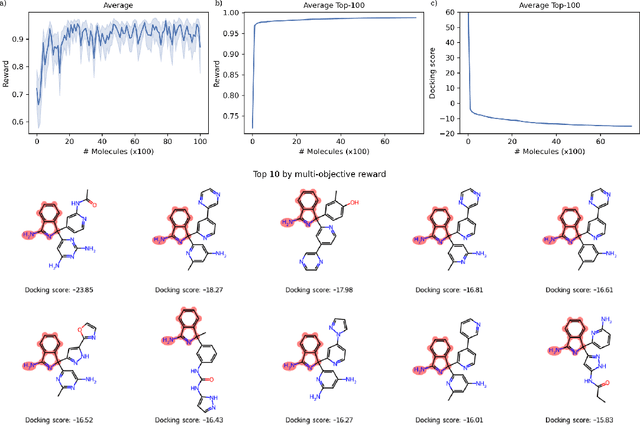
Sep 20, 2023Virtual screening of large compound libraries to identify potential hit candidates is one of the earliest steps in drug discovery. As the size of commercially available compound collections grows exponentially to the scale of billions, brute-force virtual screening using traditional tools such as docking becomes infeasible in terms of time and computational resources. Active learning and Bayesian optimization has recently been proven as effective methods of narrowing down the search space. An essential component in those methods is a surrogate machine learning model that is trained with a small subset of the library to predict the desired properties of compounds. Accurate model can achieve high sample efficiency by finding the most promising compounds with only a fraction of the whole library being virtually screened. In this study, we examined the performance of pretrained transformer-based language model and graph neural network in Bayesian optimization active learning framework. The best pretrained models identifies 58.97% of the top-50000 by docking score after screening only 0.6% of an ultra-large library containing 99.5 million compounds, improving 8% over previous state-of-the-art baseline. Through extensive benchmarks, we show that the superior performance of pretrained models persists in both structure-based and ligand-based drug discovery. Such model can serve as a boost to the accuracy and sample efficiency of active learning based molecule virtual screening.
A Transformer-based Generative Model for De Novo Molecular Design
Oct 17, 2022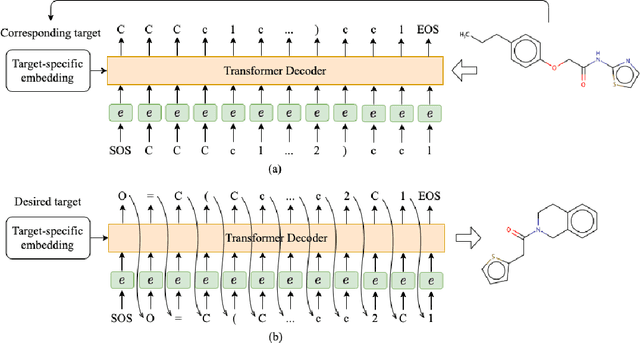
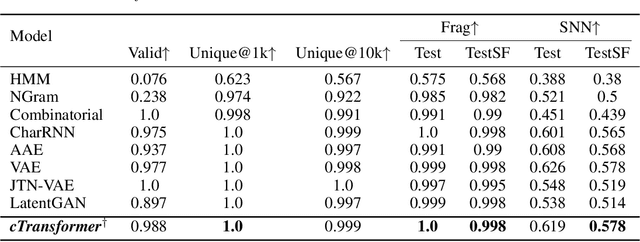
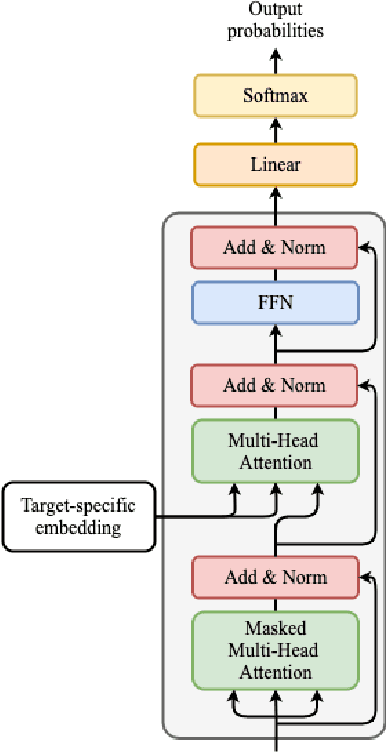
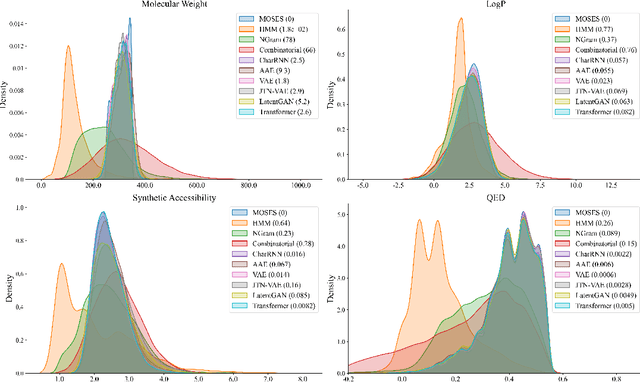
Deep learning draws a lot of attention as a new way of generating unseen structures for drug discovery. We propose a Transformer-based deep model for de novo target-specific molecular design. The proposed method is capable of generating both drug-like compounds and target-specific compounds. The latter are generated by enforcing different keys and values of the multi-head attention for each target. We allow the generation of SMILES strings to be conditional on the specified target. The sampled compounds largely occupy the real target-specific data's chemical space and also cover a significant fraction of novel compounds.