 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMulti-objective fluorescent molecule design with a data-physics dual-driven generative framework
Jan 20, 2026Designing fluorescent small molecules with tailored optical and physicochemical properties requires navigating vast, underexplored chemical space while satisfying multiple objectives and constraints. Conventional generate-score-screen approaches become impractical under such realistic design specifications, owing to their low search efficiency, unreliable generalizability of machine-learning prediction, and the prohibitive cost of quantum chemical calculation. Here we present LUMOS, a data-and-physics driven framework for inverse design of fluorescent molecules. LUMOS couples generator and predictor within a shared latent representation, enabling direct specification-to-molecule design and efficient exploration. Moreover, LUMOS combines neural networks with a fast time-dependent density functional theory (TD-DFT) calculation workflow to build a suite of complementary predictors spanning different trade-offs in speed, accuracy, and generalizability, enabling reliable property prediction across diverse scenarios. Finally, LUMOS employs a property-guided diffusion model integrated with multi-objective evolutionary algorithms, enabling de novo design and molecular optimization under multiple objectives and constraints. Across comprehensive benchmarks, LUMOS consistently outperforms baseline models in terms of accuracy, generalizability and physical plausibility for fluorescence property prediction, and demonstrates superior performance in multi-objective scaffold- and fragment-level molecular optimization. Further validation using TD-DFT and molecular dynamics (MD) simulations demonstrates that LUMOS can generate valid fluorophores that meet various target specifications. Overall, these results establish LUMOS as a data-physics dual-driven framework for general fluorophore inverse design.
Interleaved Tool-Call Reasoning for Protein Function Understanding
Jan 07, 2026Recent advances in large language models (LLMs) have highlighted the effectiveness of chain-of-thought reasoning in symbolic domains such as mathematics and programming. However, our study shows that directly transferring such text-based reasoning paradigms to protein function understanding is ineffective: reinforcement learning mainly amplifies superficial keyword patterns while failing to introduce new biological knowledge, resulting in limited generalization. We argue that protein function prediction is a knowledge-intensive scientific task that fundamentally relies on external biological priors and computational tools rather than purely internal reasoning. To address this gap, we propose PFUA, a tool-augmented protein reasoning agent that unifies problem decomposition, tool invocation, and grounded answer generation. Instead of relying on long unconstrained reasoning traces, PFUA integrates domain-specific tools to produce verifiable intermediate evidence. Experiments on four benchmarks demonstrate that PFUA consistently outperforms text-only reasoning models with an average performance improvement of 103%.
ProtTeX: Structure-In-Context Reasoning and Editing of Proteins with Large Language Models
Mar 13, 2025Large language models have made remarkable progress in the field of molecular science, particularly in understanding and generating functional small molecules. This success is largely attributed to the effectiveness of molecular tokenization strategies. In protein science, the amino acid sequence serves as the sole tokenizer for LLMs. However, many fundamental challenges in protein science are inherently structure-dependent. The absence of structure-aware tokens significantly limits the capabilities of LLMs for comprehensive biomolecular comprehension and multimodal generation. To address these challenges, we introduce a novel framework, ProtTeX, which tokenizes the protein sequences, structures, and textual information into a unified discrete space. This innovative approach enables joint training of the LLM exclusively through the Next-Token Prediction paradigm, facilitating multimodal protein reasoning and generation. ProtTeX enables general LLMs to perceive and process protein structures through sequential text input, leverage structural information as intermediate reasoning components, and generate or manipulate structures via sequential text output. Experiments demonstrate that our model achieves significant improvements in protein function prediction, outperforming the state-of-the-art domain expert model with a twofold increase in accuracy. Our framework enables high-quality conformational generation and customizable protein design. For the first time, we demonstrate that by adopting the standard training and inference pipelines from the LLM domain, ProtTeX empowers decoder-only LLMs to effectively address diverse spectrum of protein-related tasks.
ProTeX: Structure-In-Context Reasoning and Editing of Proteins with Large Language Models
Mar 11, 2025Large language models have made remarkable progress in the field of molecular science, particularly in understanding and generating functional small molecules. This success is largely attributed to the effectiveness of molecular tokenization strategies. In protein science, the amino acid sequence serves as the sole tokenizer for LLMs. However, many fundamental challenges in protein science are inherently structure-dependent. The absence of structure-aware tokens significantly limits the capabilities of LLMs for comprehensive biomolecular comprehension and multimodal generation. To address these challenges, we introduce a novel framework, ProTeX, which tokenizes the protein sequences, structures, and textual information into a unified discrete space. This innovative approach enables joint training of the LLM exclusively through the Next-Token Prediction paradigm, facilitating multimodal protein reasoning and generation. ProTeX enables general LLMs to perceive and process protein structures through sequential text input, leverage structural information as intermediate reasoning components, and generate or manipulate structures via sequential text output. Experiments demonstrate that our model achieves significant improvements in protein function prediction, outperforming the state-of-the-art domain expert model with a twofold increase in accuracy. Our framework enables high-quality conformational generation and customizable protein design. For the first time, we demonstrate that by adopting the standard training and inference pipelines from the LLM domain, ProTeX empowers decoder-only LLMs to effectively address diverse spectrum of protein-related tasks.
Generating High-Precision Force Fields for Molecular Dynamics Simulations to Study Chemical Reaction Mechanisms using Molecular Configuration Transformer
Dec 31, 2023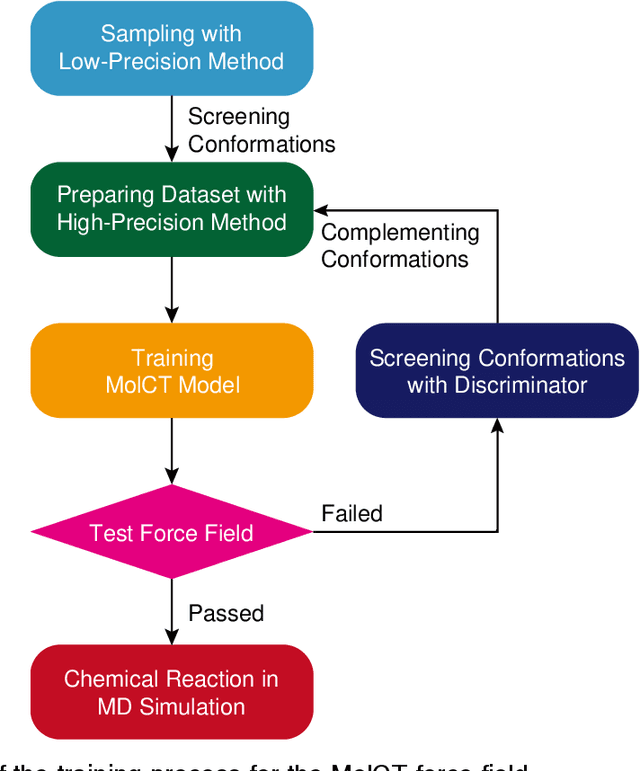
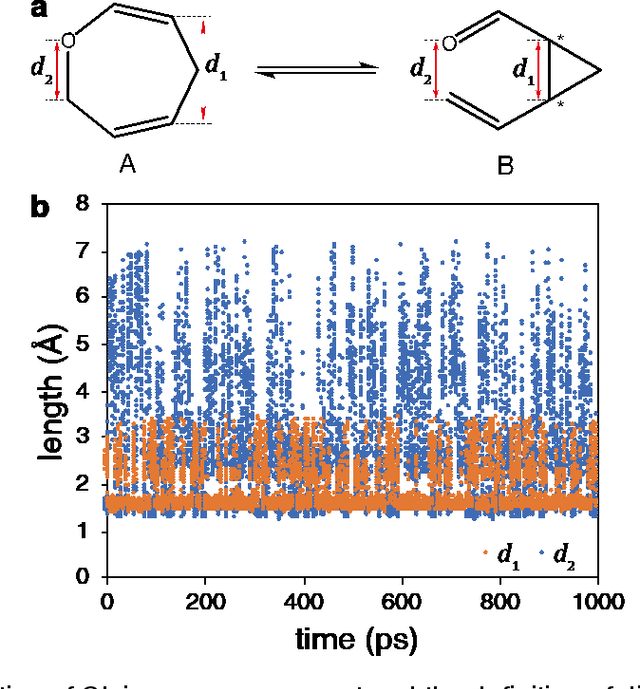
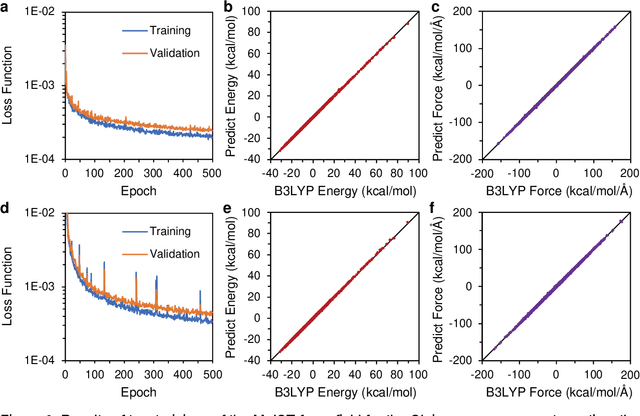
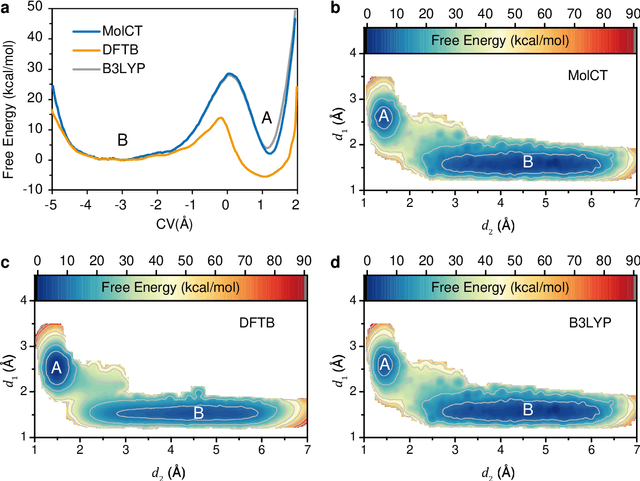
Theoretical studies on chemical reaction mechanisms have been crucial in organic chemistry. Traditionally, calculating the manually constructed molecular conformations of transition states for chemical reactions using quantum chemical calculations is the most commonly used method. However, this way is heavily dependent on individual experience and chemical intuition. In our previous study, we proposed a research paradigm that uses enhanced sampling in QM/MM molecular dynamics simulations to study chemical reactions. This approach can directly simulate the entire process of a chemical reaction. However, the computational speed limits the use of high-precision potential energy functions for simulations. To address this issue, we present a scheme for training high-precision force fields for molecular modeling using our developed graph-neural-network-based molecular model, molecular configuration transformer. This potential energy function allows for highly accurate simulations at a low computational cost, leading to more precise calculations of the mechanism of chemical reactions. We have used this approach to study a Cope rearrangement reaction and a Carbonyl insertion reaction catalyzed by Manganese. This "AI+Physics" based simulation approach is expected to become a new trend in the theoretical study of organic chemical reaction mechanisms.
Machine-Learned Invertible Coarse Graining for Multiscale Molecular Modeling
May 02, 2023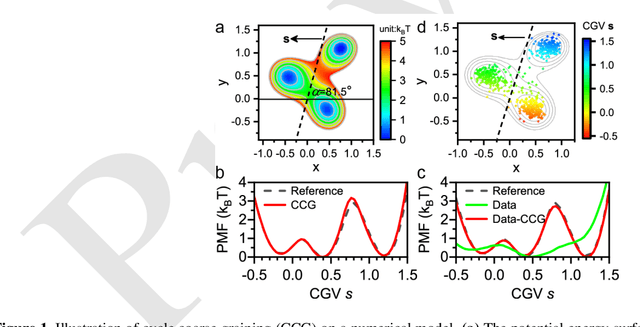
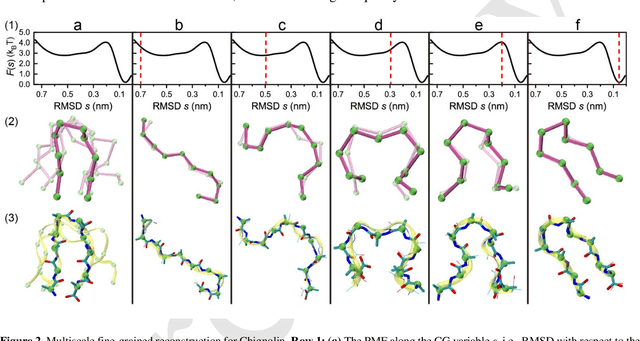
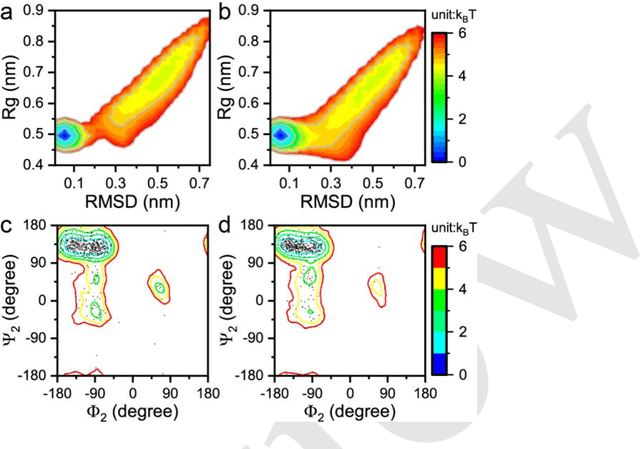
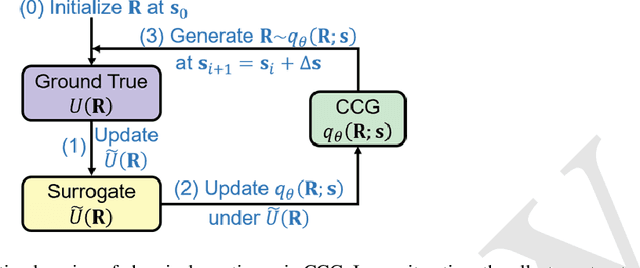
Multiscale molecular modeling is widely applied in scientific research of molecular properties over large time and length scales. Two specific challenges are commonly present in multiscale modeling, provided that information between the coarse and fine representations of molecules needs to be properly exchanged: One is to construct coarse grained (CG) models by passing information from the fine to coarse levels; the other is to restore finer molecular details given CG configurations. Although these two problems are commonly addressed independently, in this work, we present a theory connecting them, and develop a methodology called Cycle Coarse Graining (CCG) to solve both problems in a unified manner. In CCG, reconstruction can be achieved via a tractable optimization process, leading to a general method to retrieve fine details from CG simulations, which in turn, delivers a new solution to the CG problem, yielding an efficient way to calculate free energies in a rare-event-free manner. CCG thus provides a systematic way for multiscale molecular modeling, where the finer details of CG simulations can be efficiently retrieved, and the CG models can be improved consistently.
DSDP: A Blind Docking Strategy Accelerated by GPUs
Mar 16, 2023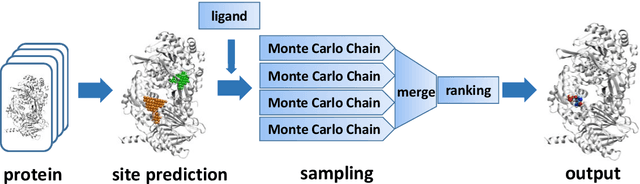
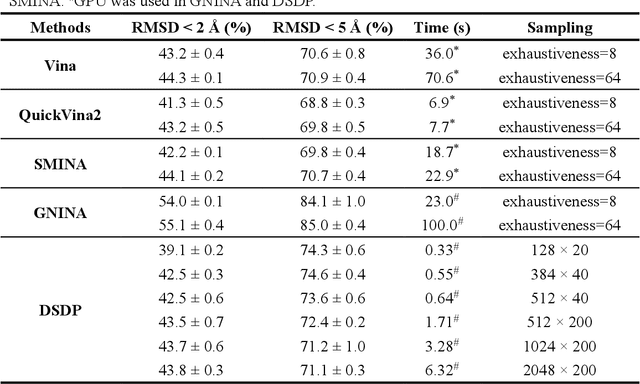
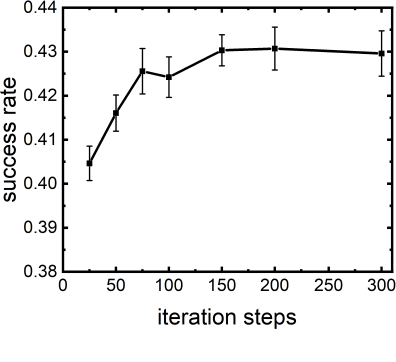
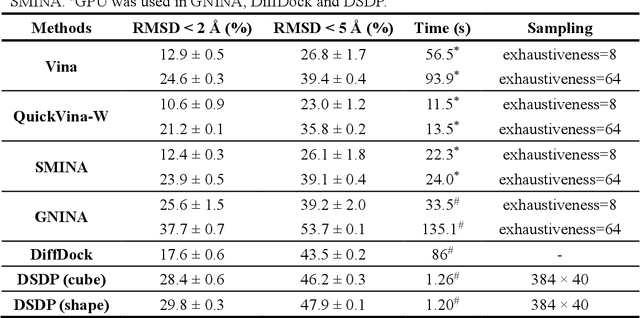
Virtual screening, including molecular docking, plays an essential role in drug discovery. Many traditional and machine-learning based methods are available to fulfil the docking task. The traditional docking methods are normally extensively time-consuming, and their performance in blind docking remains to be improved. Although the runtime of docking based on machine learning is significantly decreased, their accuracy is still limited. In this study, we take the advantage of both traditional and machine-learning based methods, and present a method Deep Site and Docking Pose (DSDP) to improve the performance of blind docking. For the traditional blind docking, the entire protein is covered by a cube, and the initial positions of ligands are randomly generated in the cube. In contract, DSDP can predict the binding site of proteins and provide an accurate searching space and initial positions for the further conformational sampling. The docking task of DSDP makes use of the score function and a similar but modified searching strategy of AutoDock Vina, accelerated by implementation in GPUs. We systematically compare its performance with the state-of-the-art methods, including Autodock Vina, GNINA, QuickVina, SMINA, and DiffDock. DSDP reaches a 29.8% top-1 success rate (RMSD < 2 {\AA}) on an unbiased and challenging test dataset with 1.2 s wall-clock computational time per system. Its performances on DUD-E dataset and the time-split PDBBind dataset used in EquiBind, TankBind, and DiffDock are also effective, presenting a 57.2% and 41.8% top-1 success rate with 0.8 s and 1.0 s per system, respectively.
Few-Shot Learning of Accurate Folding Landscape for Protein Structure Prediction
Aug 20, 2022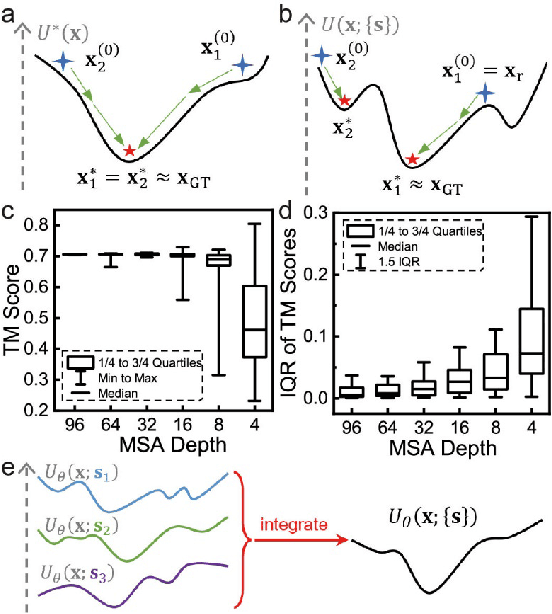
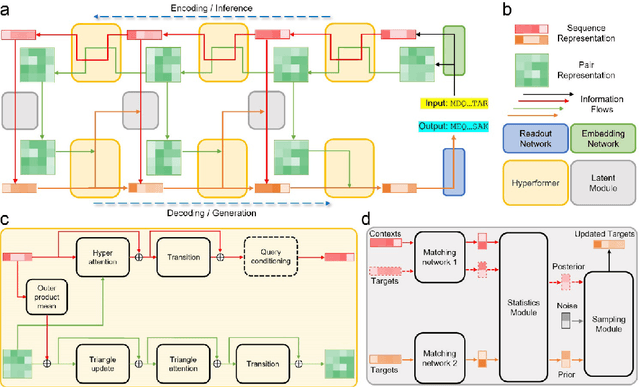
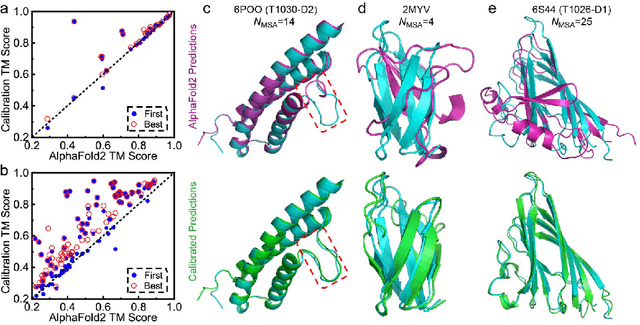
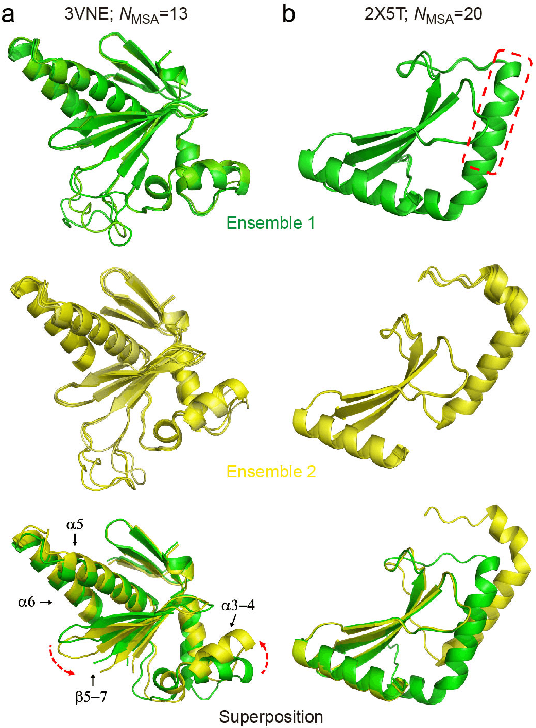
Data-driven predictive methods which can efficiently and accurately transform protein sequences into biologically active structures are highly valuable for scientific research and therapeutical development. Determining accurate folding landscape using co-evolutionary information is fundamental to the success of modern protein structure prediction methods. As the state of the art, AlphaFold2 has dramatically raised the accuracy without performing explicit co-evolutionary analysis. Nevertheless, its performance still shows strong dependence on available sequence homologs. We investigated the cause of such dependence and presented EvoGen, a meta generative model, to remedy the underperformance of AlphaFold2 for poor MSA targets. EvoGen allows us to manipulate the folding landscape either by denoising the searched MSA or by generating virtual MSA, and helps AlphaFold2 fold accurately in low-data regime or even achieve encouraging performance with single-sequence predictions. Being able to make accurate predictions with few-shot MSA not only generalizes AlphaFold2 better for orphan sequences, but also democratizes its use for high-throughput applications. Besides, EvoGen combined with AlphaFold2 yields a probabilistic structure generation method which could explore alternative conformations of protein sequences, and the task-aware differentiable algorithm for sequence generation will benefit other related tasks including protein design.
PSP: Million-level Protein Sequence Dataset for Protein Structure Prediction
Jun 24, 2022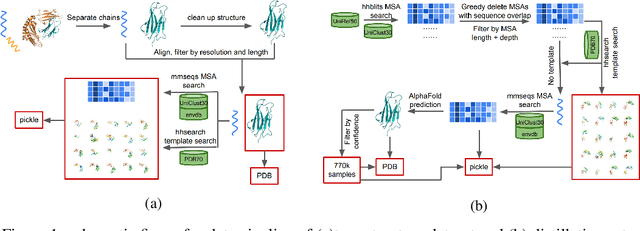
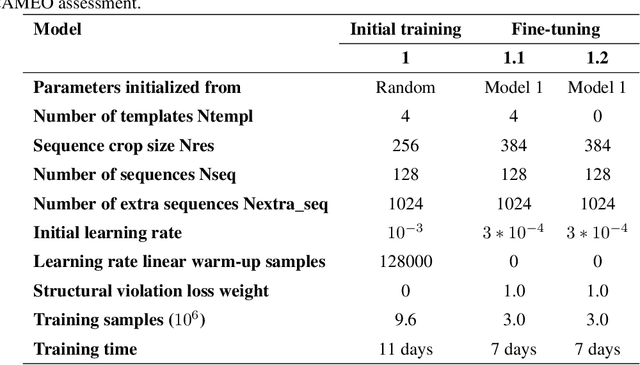
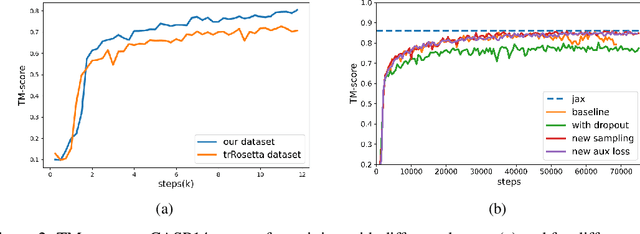
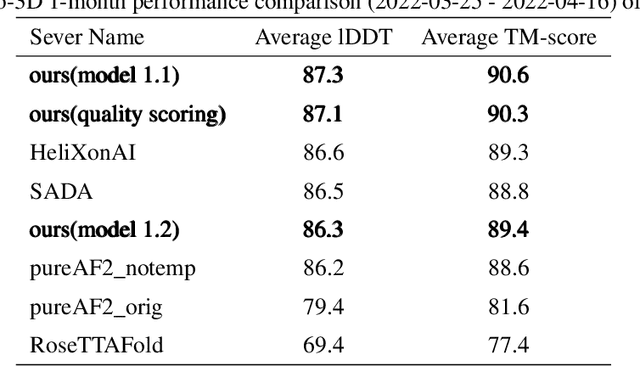
Proteins are essential component of human life and their structures are important for function and mechanism analysis. Recent work has shown the potential of AI-driven methods for protein structure prediction. However, the development of new models is restricted by the lack of dataset and benchmark training procedure. To the best of our knowledge, the existing open source datasets are far less to satisfy the needs of modern protein sequence-structure related research. To solve this problem, we present the first million-level protein structure prediction dataset with high coverage and diversity, named as PSP. This dataset consists of 570k true structure sequences (10TB) and 745k complementary distillation sequences (15TB). We provide in addition the benchmark training procedure for SOTA protein structure prediction model on this dataset. We validate the utility of this dataset for training by participating CAMEO contest in which our model won the first place. We hope our PSP dataset together with the training benchmark can enable a broader community of AI/biology researchers for AI-driven protein related research.
Molecular CT: Unifying Geometry and Representation Learning for Molecules at Different Scales
Dec 24, 2020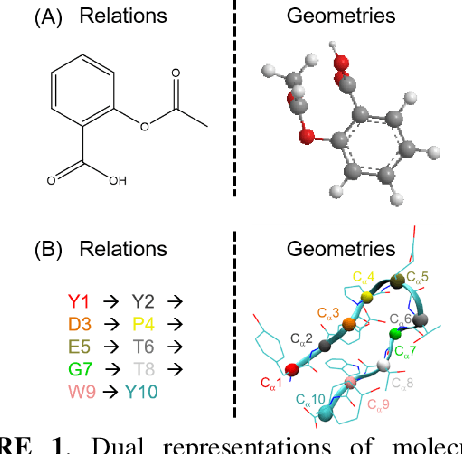
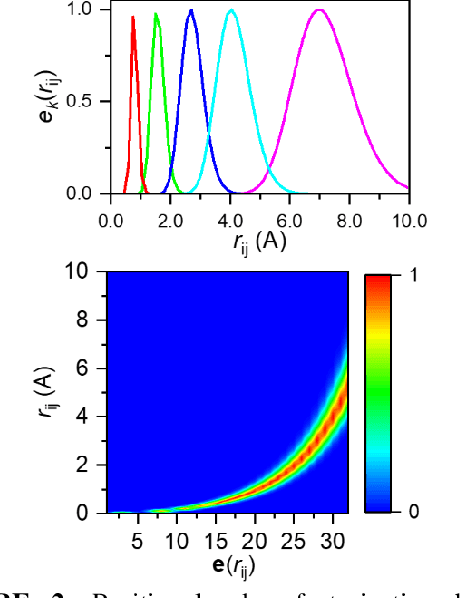
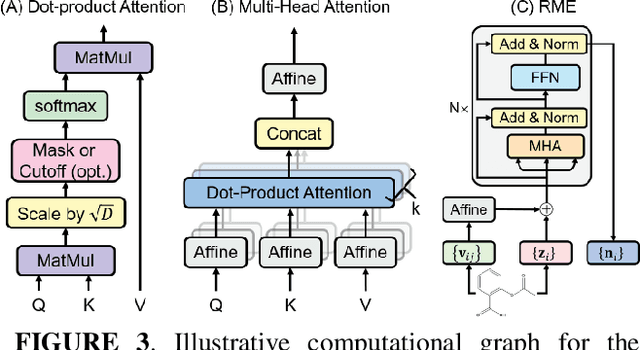
Deep learning is changing many areas in molecular physics, and it has shown great potential to deliver new solutions to challenging molecular modeling problems. Along with this trend arises the increasing demand of expressive and versatile neural network architectures which are compatible with molecular systems. A new deep neural network architecture, Molecular Configuration Transformer (Molecular CT), is introduced for this purpose. Molecular CT is composed of a relation-aware encoder module and a computationally universal geometry learning unit, thus able to account for the relational constraints between particles meanwhile scalable to different particle numbers and invariant w.r.t. the trans-rotational transforms. The computational efficiency and universality make Molecular CT versatile for a variety of molecular learning scenarios and especially appealing for transferable representation learning across different molecular systems. As examples, we show that Molecular CT enables representational learning for molecular systems at different scales, and achieves comparable or improved results on common benchmarks using a more light-weighted structure compared to baseline models.