 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDSDP: A Blind Docking Strategy Accelerated by GPUs
Mar 16, 2023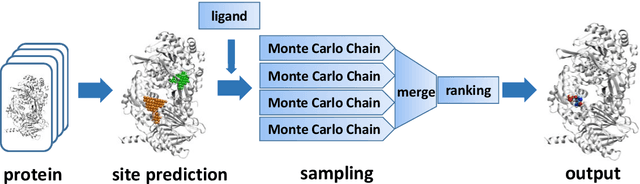
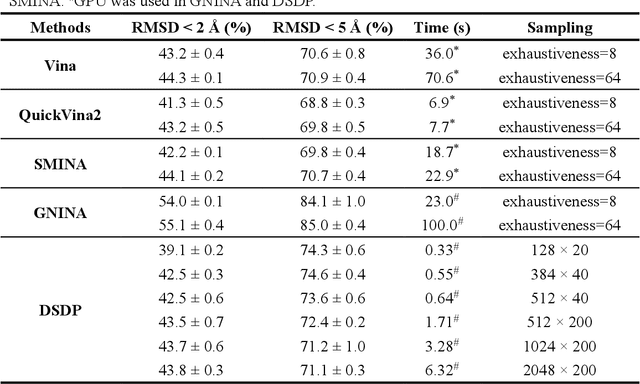
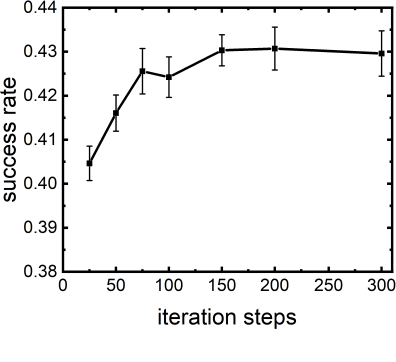
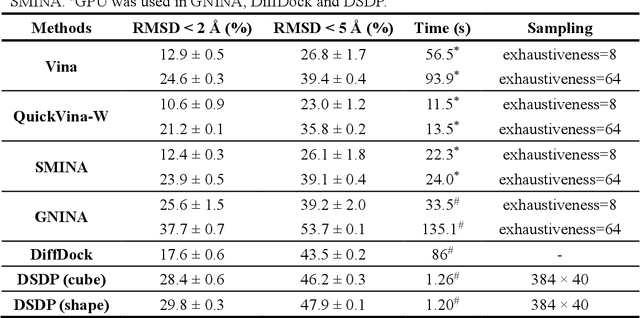
Virtual screening, including molecular docking, plays an essential role in drug discovery. Many traditional and machine-learning based methods are available to fulfil the docking task. The traditional docking methods are normally extensively time-consuming, and their performance in blind docking remains to be improved. Although the runtime of docking based on machine learning is significantly decreased, their accuracy is still limited. In this study, we take the advantage of both traditional and machine-learning based methods, and present a method Deep Site and Docking Pose (DSDP) to improve the performance of blind docking. For the traditional blind docking, the entire protein is covered by a cube, and the initial positions of ligands are randomly generated in the cube. In contract, DSDP can predict the binding site of proteins and provide an accurate searching space and initial positions for the further conformational sampling. The docking task of DSDP makes use of the score function and a similar but modified searching strategy of AutoDock Vina, accelerated by implementation in GPUs. We systematically compare its performance with the state-of-the-art methods, including Autodock Vina, GNINA, QuickVina, SMINA, and DiffDock. DSDP reaches a 29.8% top-1 success rate (RMSD < 2 {\AA}) on an unbiased and challenging test dataset with 1.2 s wall-clock computational time per system. Its performances on DUD-E dataset and the time-split PDBBind dataset used in EquiBind, TankBind, and DiffDock are also effective, presenting a 57.2% and 41.8% top-1 success rate with 0.8 s and 1.0 s per system, respectively.