 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFew-Shot Learning of Accurate Folding Landscape for Protein Structure Prediction
Aug 20, 2022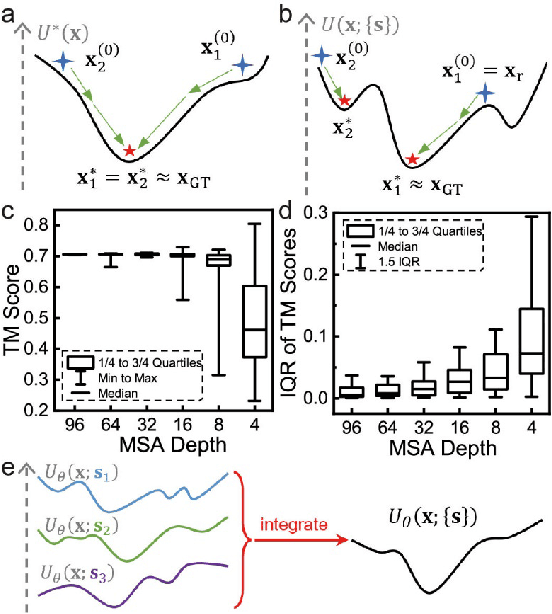
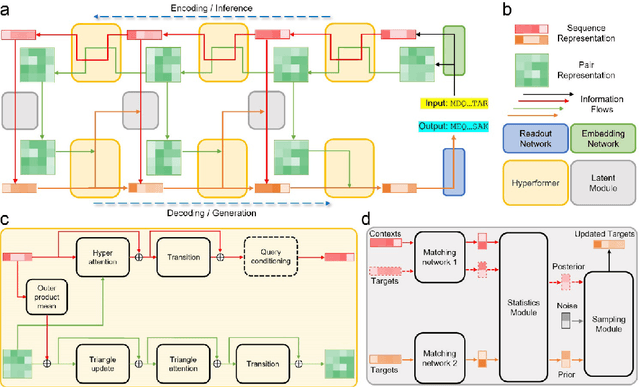
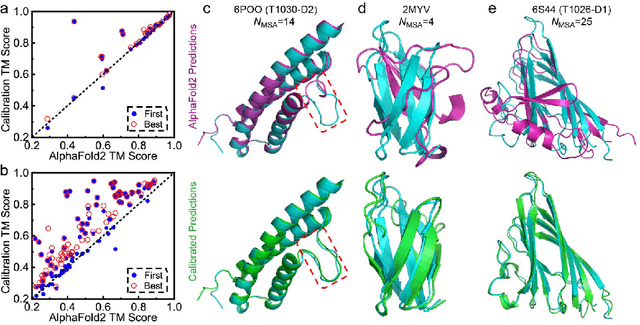
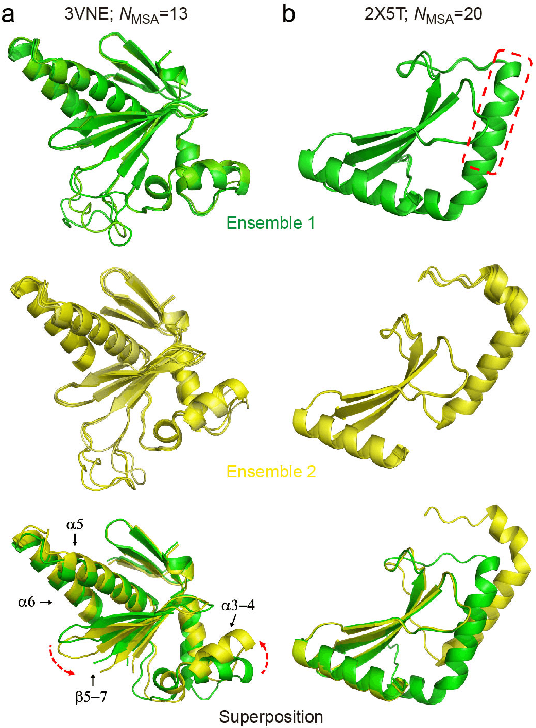
Data-driven predictive methods which can efficiently and accurately transform protein sequences into biologically active structures are highly valuable for scientific research and therapeutical development. Determining accurate folding landscape using co-evolutionary information is fundamental to the success of modern protein structure prediction methods. As the state of the art, AlphaFold2 has dramatically raised the accuracy without performing explicit co-evolutionary analysis. Nevertheless, its performance still shows strong dependence on available sequence homologs. We investigated the cause of such dependence and presented EvoGen, a meta generative model, to remedy the underperformance of AlphaFold2 for poor MSA targets. EvoGen allows us to manipulate the folding landscape either by denoising the searched MSA or by generating virtual MSA, and helps AlphaFold2 fold accurately in low-data regime or even achieve encouraging performance with single-sequence predictions. Being able to make accurate predictions with few-shot MSA not only generalizes AlphaFold2 better for orphan sequences, but also democratizes its use for high-throughput applications. Besides, EvoGen combined with AlphaFold2 yields a probabilistic structure generation method which could explore alternative conformations of protein sequences, and the task-aware differentiable algorithm for sequence generation will benefit other related tasks including protein design.
PSP: Million-level Protein Sequence Dataset for Protein Structure Prediction
Jun 24, 2022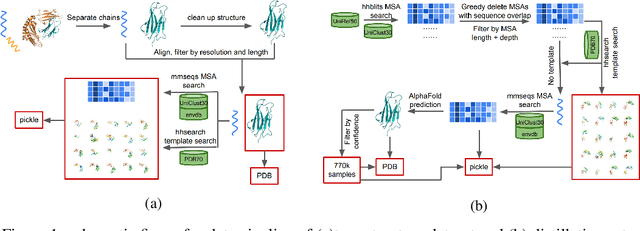
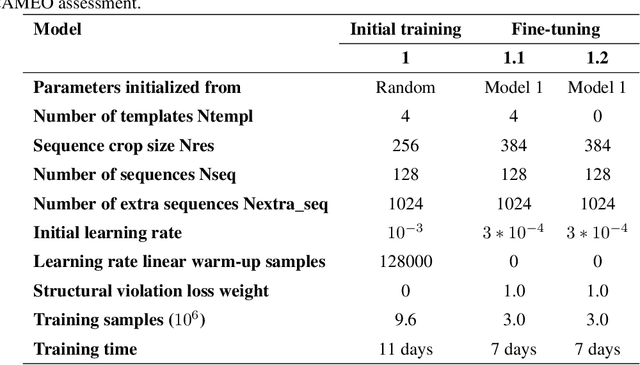
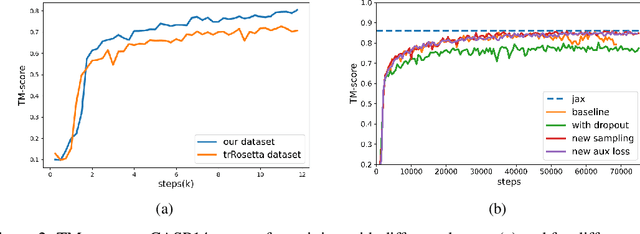
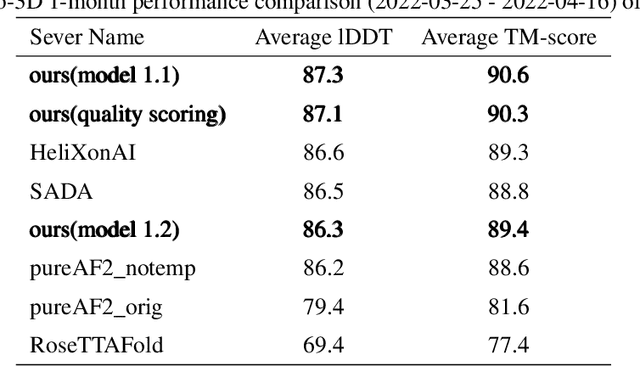
Proteins are essential component of human life and their structures are important for function and mechanism analysis. Recent work has shown the potential of AI-driven methods for protein structure prediction. However, the development of new models is restricted by the lack of dataset and benchmark training procedure. To the best of our knowledge, the existing open source datasets are far less to satisfy the needs of modern protein sequence-structure related research. To solve this problem, we present the first million-level protein structure prediction dataset with high coverage and diversity, named as PSP. This dataset consists of 570k true structure sequences (10TB) and 745k complementary distillation sequences (15TB). We provide in addition the benchmark training procedure for SOTA protein structure prediction model on this dataset. We validate the utility of this dataset for training by participating CAMEO contest in which our model won the first place. We hope our PSP dataset together with the training benchmark can enable a broader community of AI/biology researchers for AI-driven protein related research.