 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSpatial-ORMLLM: Improve Spatial Relation Understanding in the Operating Room with Multimodal Large Language Model
Aug 11, 2025Precise spatial modeling in the operating room (OR) is foundational to many clinical tasks, supporting intraoperative awareness, hazard avoidance, and surgical decision-making. While existing approaches leverage large-scale multimodal datasets for latent-space alignment to implicitly learn spatial relationships, they overlook the 3D capabilities of MLLMs. However, this approach raises two issues: (1) Operating rooms typically lack multiple video and audio sensors, making multimodal 3D data difficult to obtain; (2) Training solely on readily available 2D data fails to capture fine-grained details in complex scenes. To address this gap, we introduce Spatial-ORMLLM, the first large vision-language model for 3D spatial reasoning in operating rooms using only RGB modality to infer volumetric and semantic cues, enabling downstream medical tasks with detailed and holistic spatial context. Spatial-ORMLLM incorporates a Spatial-Enhanced Feature Fusion Block, which integrates 2D modality inputs with rich 3D spatial knowledge extracted by the estimation algorithm and then feeds the combined features into the visual tower. By employing a unified end-to-end MLLM framework, it combines powerful spatial features with textual features to deliver robust 3D scene reasoning without any additional expert annotations or sensor inputs. Experiments on multiple benchmark clinical datasets demonstrate that Spatial-ORMLLM achieves state-of-the-art performance and generalizes robustly to previously unseen surgical scenarios and downstream tasks.
SMILE: a Scale-aware Multiple Instance Learning Method for Multicenter STAS Lung Cancer Histopathology Diagnosis
Mar 18, 2025Spread through air spaces (STAS) represents a newly identified aggressive pattern in lung cancer, which is known to be associated with adverse prognostic factors and complex pathological features. Pathologists currently rely on time consuming manual assessments, which are highly subjective and prone to variation. This highlights the urgent need for automated and precise diag nostic solutions. 2,970 lung cancer tissue slides are comprised from multiple centers, re-diagnosed them, and constructed and publicly released three lung cancer STAS datasets: STAS CSU (hospital), STAS TCGA, and STAS CPTAC. All STAS datasets provide corresponding pathological feature diagnoses and related clinical data. To address the bias, sparse and heterogeneous nature of STAS, we propose an scale-aware multiple instance learning(SMILE) method for STAS diagnosis of lung cancer. By introducing a scale-adaptive attention mechanism, the SMILE can adaptively adjust high attention instances, reducing over-reliance on local regions and promoting consistent detection of STAS lesions. Extensive experiments show that SMILE achieved competitive diagnostic results on STAS CSU, diagnosing 251 and 319 STAS samples in CPTAC andTCGA,respectively, surpassing clinical average AUC. The 11 open baseline results are the first to be established for STAS research, laying the foundation for the future expansion, interpretability, and clinical integration of computational pathology technologies. The datasets and code are available at https://anonymous.4open.science/r/IJCAI25-1DA1.
Feature-interactive Siamese graph encoder-based image analysis to predict STAS from histopathology images in lung cancer
Nov 22, 2024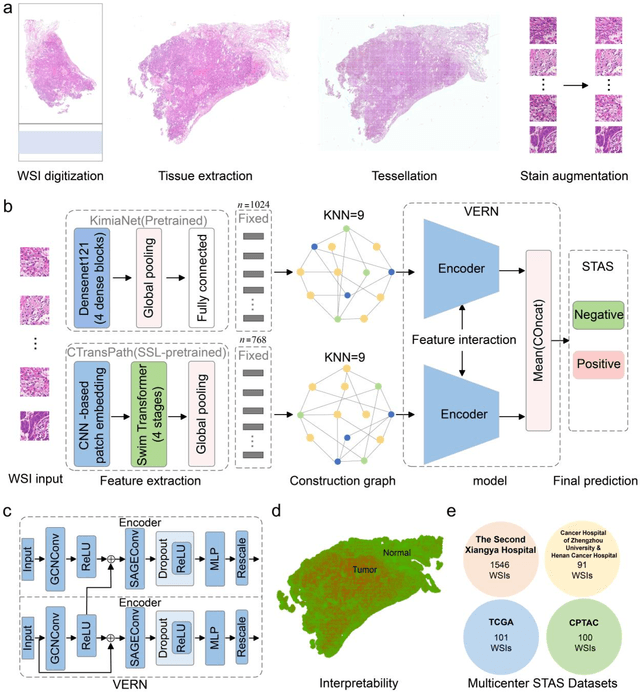
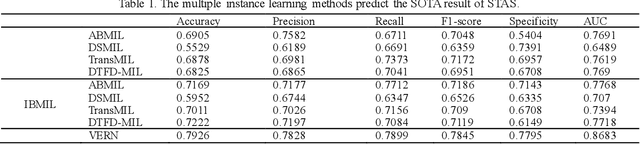
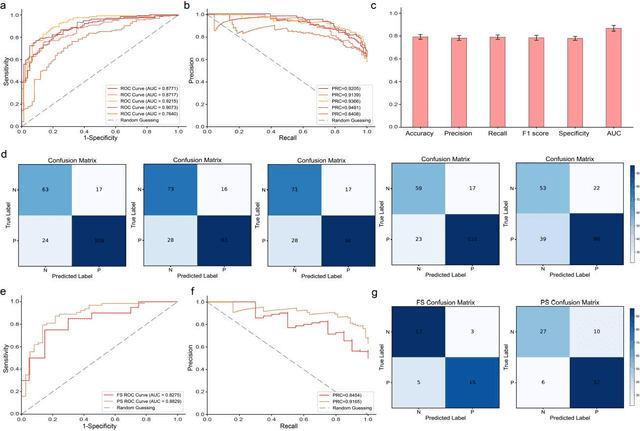
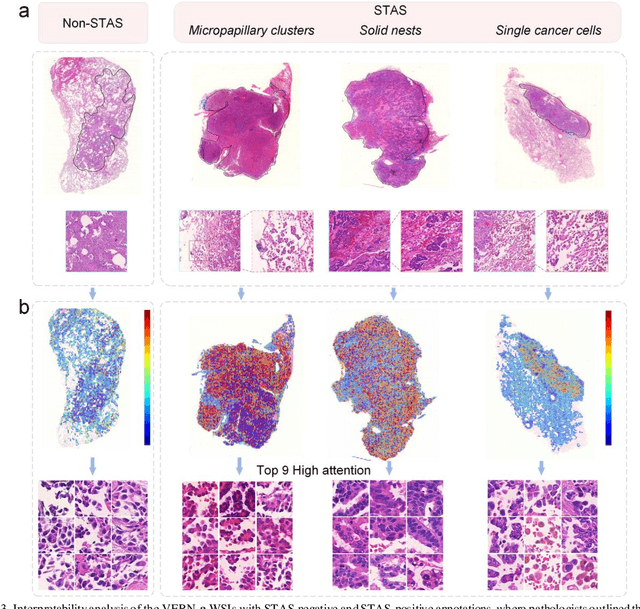
Spread through air spaces (STAS) is a distinct invasion pattern in lung cancer, crucial for prognosis assessment and guiding surgical decisions. Histopathology is the gold standard for STAS detection, yet traditional methods are subjective, time-consuming, and prone to misdiagnosis, limiting large-scale applications. We present VERN, an image analysis model utilizing a feature-interactive Siamese graph encoder to predict STAS from lung cancer histopathological images. VERN captures spatial topological features with feature sharing and skip connections to enhance model training. Using 1,546 histopathology slides, we built a large single-cohort STAS lung cancer dataset. VERN achieved an AUC of 0.9215 in internal validation and AUCs of 0.8275 and 0.8829 in frozen and paraffin-embedded test sections, respectively, demonstrating clinical-grade performance. Validated on a single-cohort and three external datasets, VERN showed robust predictive performance and generalizability, providing an open platform (http://plr.20210706.xyz:5000/) to enhance STAS diagnosis efficiency and accuracy.
FedDP: Privacy-preserving method based on federated learning for histopathology image segmentation
Nov 07, 2024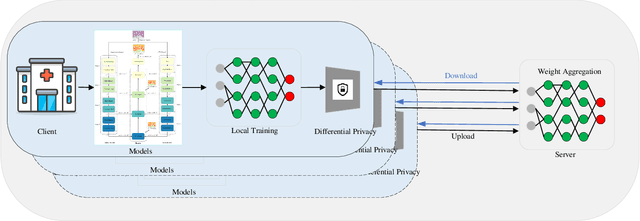

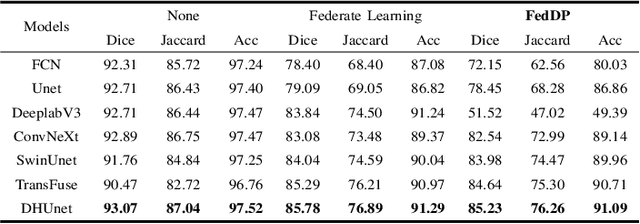
Hematoxylin and Eosin (H&E) staining of whole slide images (WSIs) is considered the gold standard for pathologists and medical practitioners for tumor diagnosis, surgical planning, and post-operative assessment. With the rapid advancement of deep learning technologies, the development of numerous models based on convolutional neural networks and transformer-based models has been applied to the precise segmentation of WSIs. However, due to privacy regulations and the need to protect patient confidentiality, centralized storage and processing of image data are impractical. Training a centralized model directly is challenging to implement in medical settings due to these privacy concerns.This paper addresses the dispersed nature and privacy sensitivity of medical image data by employing a federated learning framework, allowing medical institutions to collaboratively learn while protecting patient privacy. Additionally, to address the issue of original data reconstruction through gradient inversion during the federated learning training process, differential privacy introduces noise into the model updates, preventing attackers from inferring the contributions of individual samples, thereby protecting the privacy of the training data.Experimental results show that the proposed method, FedDP, minimally impacts model accuracy while effectively safeguarding the privacy of cancer pathology image data, with only a slight decrease in Dice, Jaccard, and Acc indices by 0.55%, 0.63%, and 0.42%, respectively. This approach facilitates cross-institutional collaboration and knowledge sharing while protecting sensitive data privacy, providing a viable solution for further research and application in the medical field.
FORESEE: Multimodal and Multi-view Representation Learning for Robust Prediction of Cancer Survival
May 13, 2024Integrating the different data modalities of cancer patients can significantly improve the predictive performance of patient survival. However, most existing methods ignore the simultaneous utilization of rich semantic features at different scales in pathology images. When collecting multimodal data and extracting features, there is a likelihood of encountering intra-modality missing data, introducing noise into the multimodal data. To address these challenges, this paper proposes a new end-to-end framework, FORESEE, for robustly predicting patient survival by mining multimodal information. Specifically, the cross-fusion transformer effectively utilizes features at the cellular level, tissue level, and tumor heterogeneity level to correlate prognosis through a cross-scale feature cross-fusion method. This enhances the ability of pathological image feature representation. Secondly, the hybrid attention encoder (HAE) uses the denoising contextual attention module to obtain the contextual relationship features and local detail features of the molecular data. HAE's channel attention module obtains global features of molecular data. Furthermore, to address the issue of missing information within modalities, we propose an asymmetrically masked triplet masked autoencoder to reconstruct lost information within modalities. Extensive experiments demonstrate the superiority of our method over state-of-the-art methods on four benchmark datasets in both complete and missing settings.
Opportunities and challenges in the application of large artificial intelligence models in radiology
Mar 24, 2024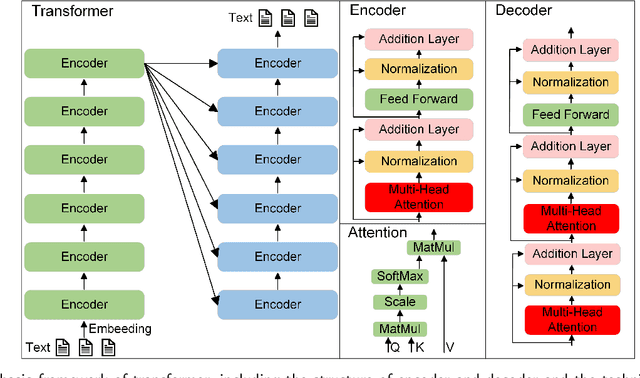
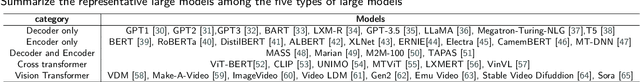
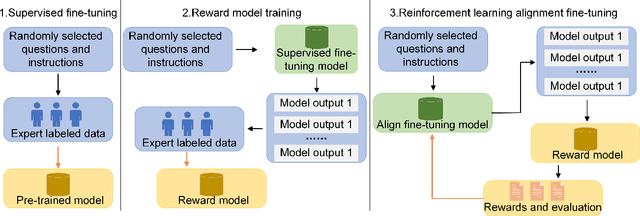
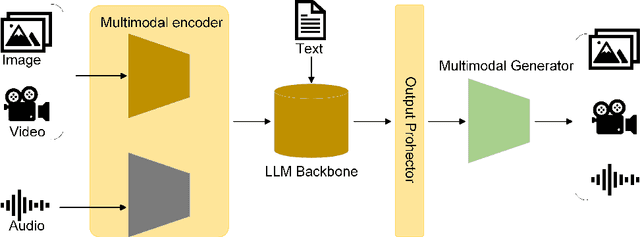
Influenced by ChatGPT, artificial intelligence (AI) large models have witnessed a global upsurge in large model research and development. As people enjoy the convenience by this AI large model, more and more large models in subdivided fields are gradually being proposed, especially large models in radiology imaging field. This article first introduces the development history of large models, technical details, workflow, working principles of multimodal large models and working principles of video generation large models. Secondly, we summarize the latest research progress of AI large models in radiology education, radiology report generation, applications of unimodal and multimodal radiology. Finally, this paper also summarizes some of the challenges of large AI models in radiology, with the aim of better promoting the rapid revolution in the field of radiography.
SELECTOR: Heterogeneous graph network with convolutional masked autoencoder for multimodal robust prediction of cancer survival
Mar 14, 2024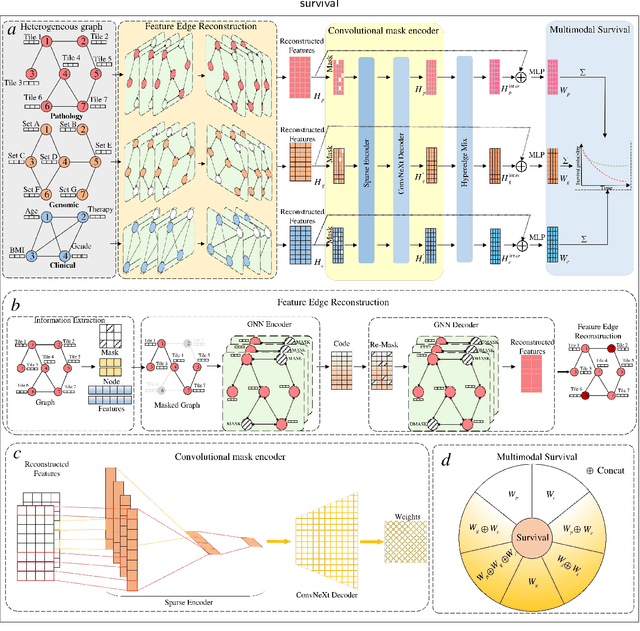
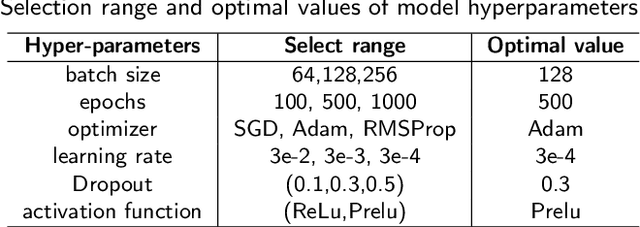
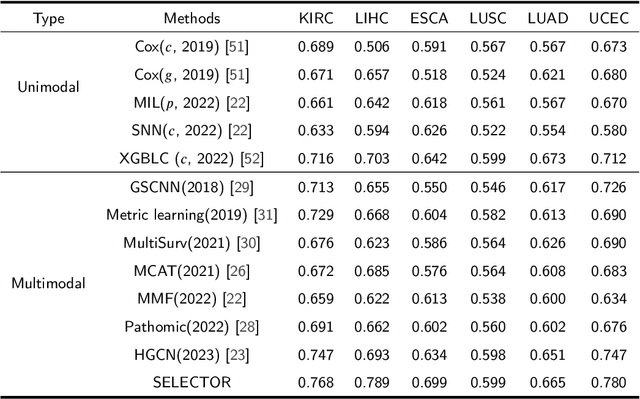
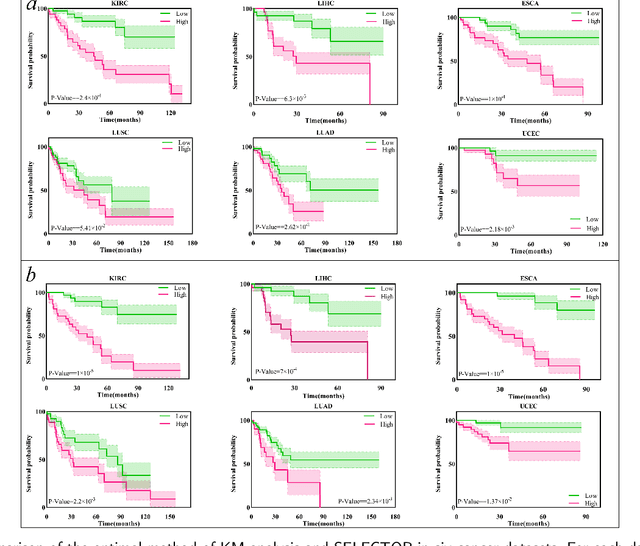
Accurately predicting the survival rate of cancer patients is crucial for aiding clinicians in planning appropriate treatment, reducing cancer-related medical expenses, and significantly enhancing patients' quality of life. Multimodal prediction of cancer patient survival offers a more comprehensive and precise approach. However, existing methods still grapple with challenges related to missing multimodal data and information interaction within modalities. This paper introduces SELECTOR, a heterogeneous graph-aware network based on convolutional mask encoders for robust multimodal prediction of cancer patient survival. SELECTOR comprises feature edge reconstruction, convolutional mask encoder, feature cross-fusion, and multimodal survival prediction modules. Initially, we construct a multimodal heterogeneous graph and employ the meta-path method for feature edge reconstruction, ensuring comprehensive incorporation of feature information from graph edges and effective embedding of nodes. To mitigate the impact of missing features within the modality on prediction accuracy, we devised a convolutional masked autoencoder (CMAE) to process the heterogeneous graph post-feature reconstruction. Subsequently, the feature cross-fusion module facilitates communication between modalities, ensuring that output features encompass all features of the modality and relevant information from other modalities. Extensive experiments and analysis on six cancer datasets from TCGA demonstrate that our method significantly outperforms state-of-the-art methods in both modality-missing and intra-modality information-confirmed cases. Our codes are made available at https://github.com/panliangrui/Selector.
ESM-NBR: fast and accurate nucleic acid-binding residue prediction via protein language model feature representation and multi-task learning
Dec 01, 2023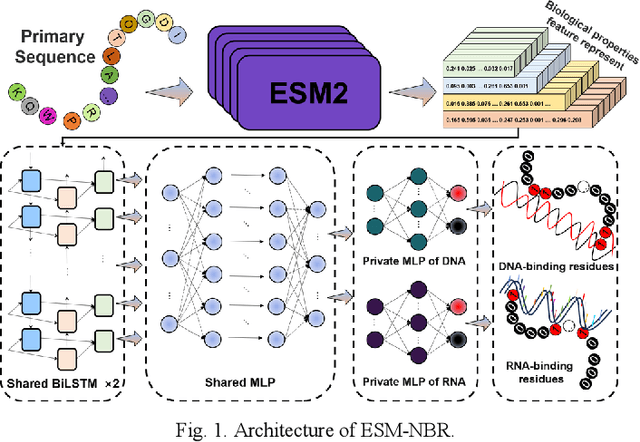
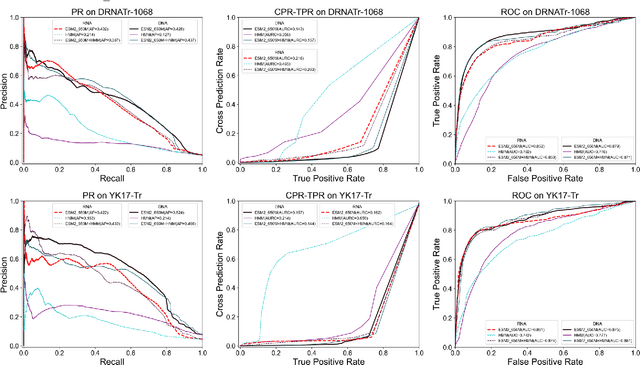
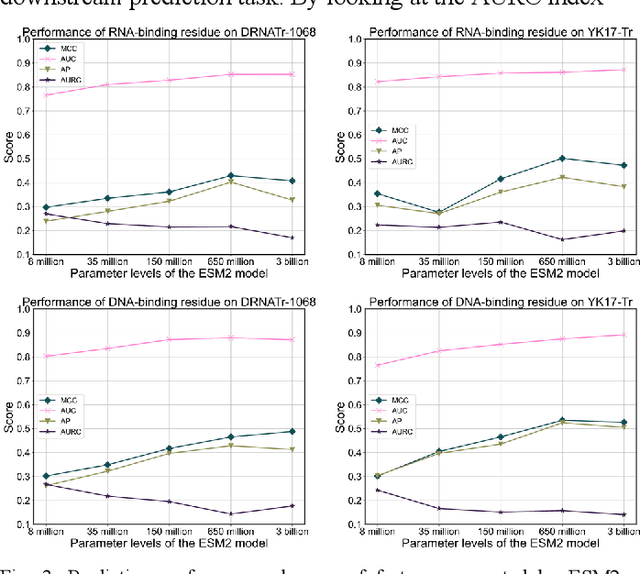
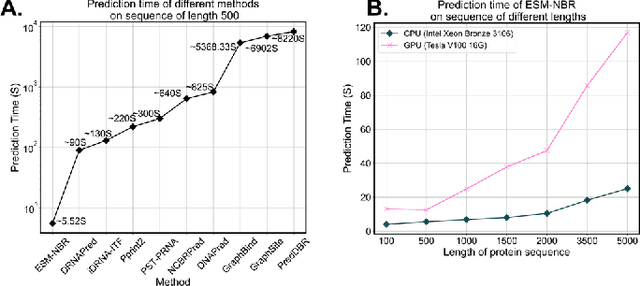
Protein-nucleic acid interactions play a very important role in a variety of biological activities. Accurate identification of nucleic acid-binding residues is a critical step in understanding the interaction mechanisms. Although many computationally based methods have been developed to predict nucleic acid-binding residues, challenges remain. In this study, a fast and accurate sequence-based method, called ESM-NBR, is proposed. In ESM-NBR, we first use the large protein language model ESM2 to extract discriminative biological properties feature representation from protein primary sequences; then, a multi-task deep learning model composed of stacked bidirectional long short-term memory (BiLSTM) and multi-layer perceptron (MLP) networks is employed to explore common and private information of DNA- and RNA-binding residues with ESM2 feature as input. Experimental results on benchmark data sets demonstrate that the prediction performance of ESM2 feature representation comprehensively outperforms evolutionary information-based hidden Markov model (HMM) features. Meanwhile, the ESM-NBR obtains the MCC values for DNA-binding residues prediction of 0.427 and 0.391 on two independent test sets, which are 18.61 and 10.45% higher than those of the second-best methods, respectively. Moreover, by completely discarding the time-cost multiple sequence alignment process, the prediction speed of ESM-NBR far exceeds that of existing methods (5.52s for a protein sequence of length 500, which is about 16 times faster than the second-fastest method). A user-friendly standalone package and the data of ESM-NBR are freely available for academic use at: https://github.com/wwzll123/ESM-NBR.
LDCSF: Local depth convolution-based Swim framework for classifying multi-label histopathology images
Aug 21, 2023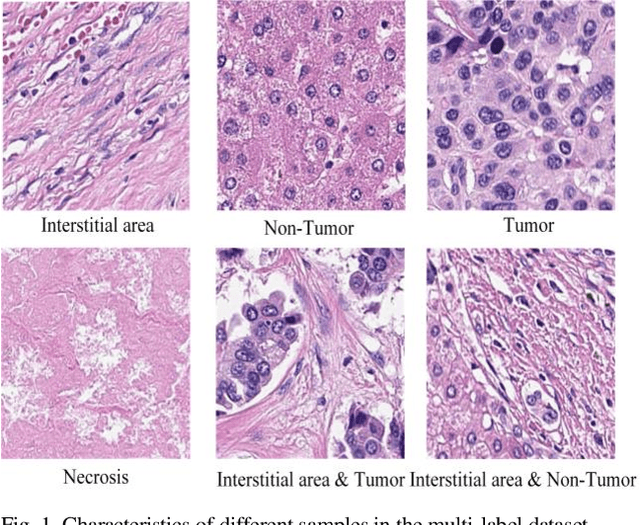
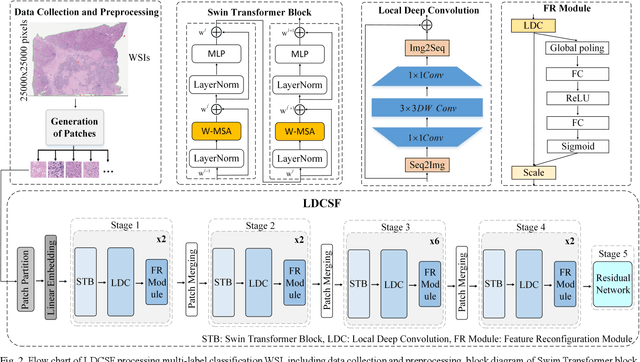
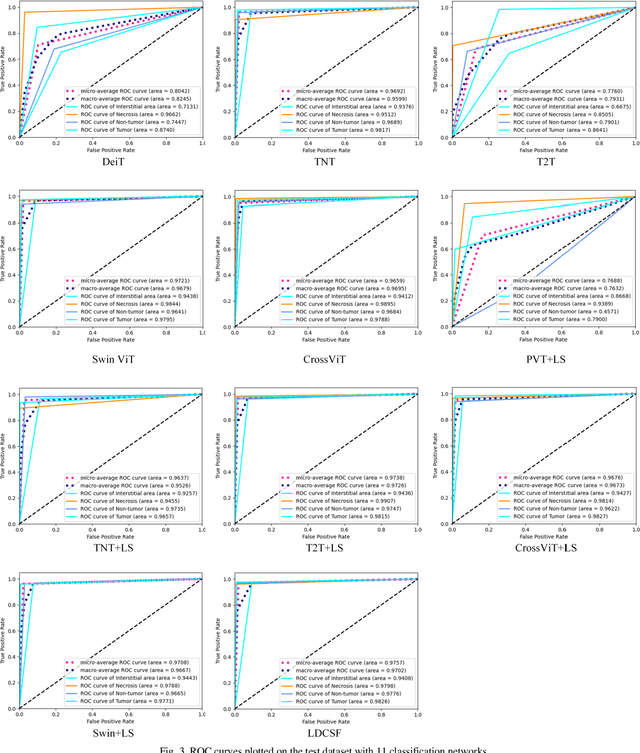
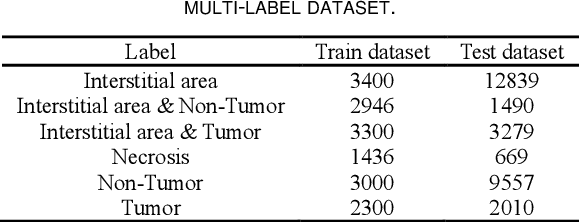
Histopathological images are the gold standard for diagnosing liver cancer. However, the accuracy of fully digital diagnosis in computational pathology needs to be improved. In this paper, in order to solve the problem of multi-label and low classification accuracy of histopathology images, we propose a locally deep convolutional Swim framework (LDCSF) to classify multi-label histopathology images. In order to be able to provide local field of view diagnostic results, we propose the LDCSF model, which consists of a Swin transformer module, a local depth convolution (LDC) module, a feature reconstruction (FR) module, and a ResNet module. The Swin transformer module reduces the amount of computation generated by the attention mechanism by limiting the attention to each window. The LDC then reconstructs the attention map and performs convolution operations in multiple channels, passing the resulting feature map to the next layer. The FR module uses the corresponding weight coefficient vectors obtained from the channels to dot product with the original feature map vector matrix to generate representative feature maps. Finally, the residual network undertakes the final classification task. As a result, the classification accuracy of LDCSF for interstitial area, necrosis, non-tumor and tumor reached 0.9460, 0.9960, 0.9808, 0.9847, respectively. Finally, we use the results of multi-label pathological image classification to calculate the tumor-to-stromal ratio, which lays the foundation for the analysis of the microenvironment of liver cancer histopathological images. Second, we released a multilabel histopathology image of liver cancer, our code and data are available at https://github.com/panliangrui/LSF.
PACS: Prediction and analysis of cancer subtypes from multi-omics data based on a multi-head attention mechanism model
Aug 21, 2023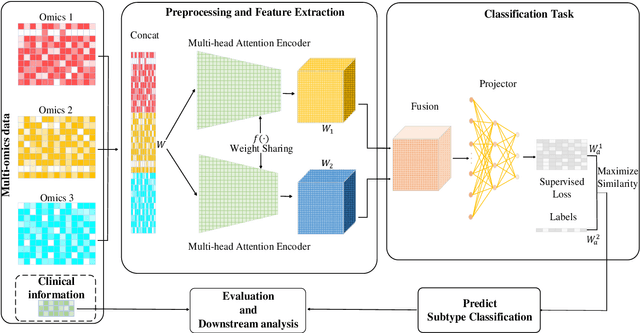
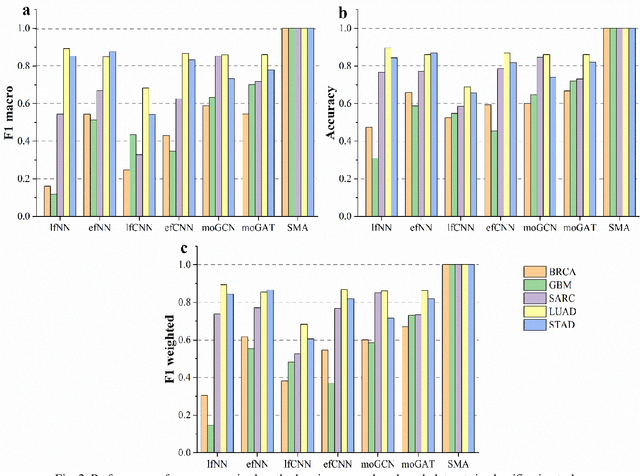
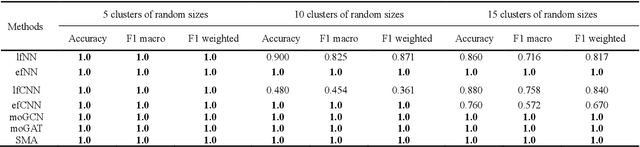
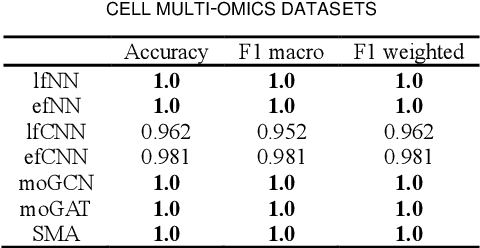
Due to the high heterogeneity and clinical characteristics of cancer, there are significant differences in multi-omic data and clinical characteristics among different cancer subtypes. Therefore, accurate classification of cancer subtypes can help doctors choose the most appropriate treatment options, improve treatment outcomes, and provide more accurate patient survival predictions. In this study, we propose a supervised multi-head attention mechanism model (SMA) to classify cancer subtypes successfully. The attention mechanism and feature sharing module of the SMA model can successfully learn the global and local feature information of multi-omics data. Second, it enriches the parameters of the model by deeply fusing multi-head attention encoders from Siamese through the fusion module. Validated by extensive experiments, the SMA model achieves the highest accuracy, F1 macroscopic, F1 weighted, and accurate classification of cancer subtypes in simulated, single-cell, and cancer multiomics datasets compared to AE, CNN, and GNN-based models. Therefore, we contribute to future research on multiomics data using our attention-based approach.