 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeESM-NBR: fast and accurate nucleic acid-binding residue prediction via protein language model feature representation and multi-task learning
Dec 01, 2023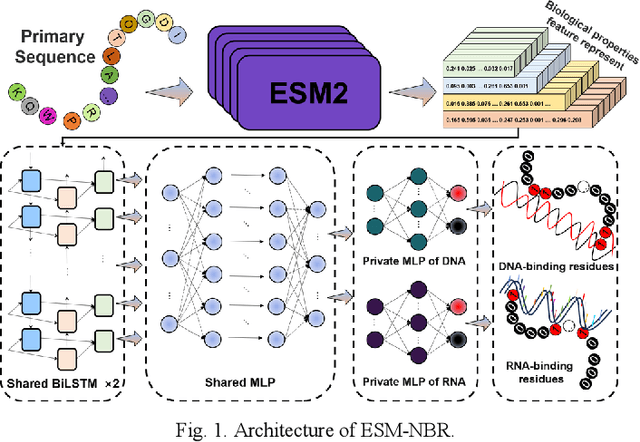
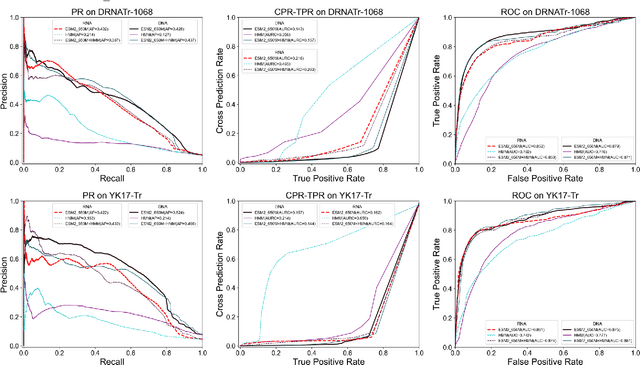
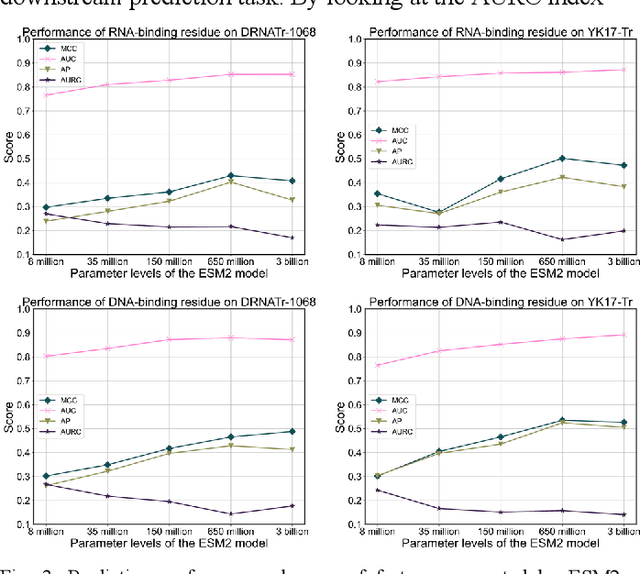
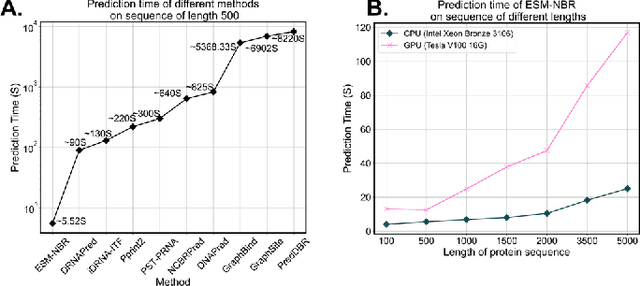
Protein-nucleic acid interactions play a very important role in a variety of biological activities. Accurate identification of nucleic acid-binding residues is a critical step in understanding the interaction mechanisms. Although many computationally based methods have been developed to predict nucleic acid-binding residues, challenges remain. In this study, a fast and accurate sequence-based method, called ESM-NBR, is proposed. In ESM-NBR, we first use the large protein language model ESM2 to extract discriminative biological properties feature representation from protein primary sequences; then, a multi-task deep learning model composed of stacked bidirectional long short-term memory (BiLSTM) and multi-layer perceptron (MLP) networks is employed to explore common and private information of DNA- and RNA-binding residues with ESM2 feature as input. Experimental results on benchmark data sets demonstrate that the prediction performance of ESM2 feature representation comprehensively outperforms evolutionary information-based hidden Markov model (HMM) features. Meanwhile, the ESM-NBR obtains the MCC values for DNA-binding residues prediction of 0.427 and 0.391 on two independent test sets, which are 18.61 and 10.45% higher than those of the second-best methods, respectively. Moreover, by completely discarding the time-cost multiple sequence alignment process, the prediction speed of ESM-NBR far exceeds that of existing methods (5.52s for a protein sequence of length 500, which is about 16 times faster than the second-fastest method). A user-friendly standalone package and the data of ESM-NBR are freely available for academic use at: https://github.com/wwzll123/ESM-NBR.