 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSELECTOR: Heterogeneous graph network with convolutional masked autoencoder for multimodal robust prediction of cancer survival
Mar 14, 2024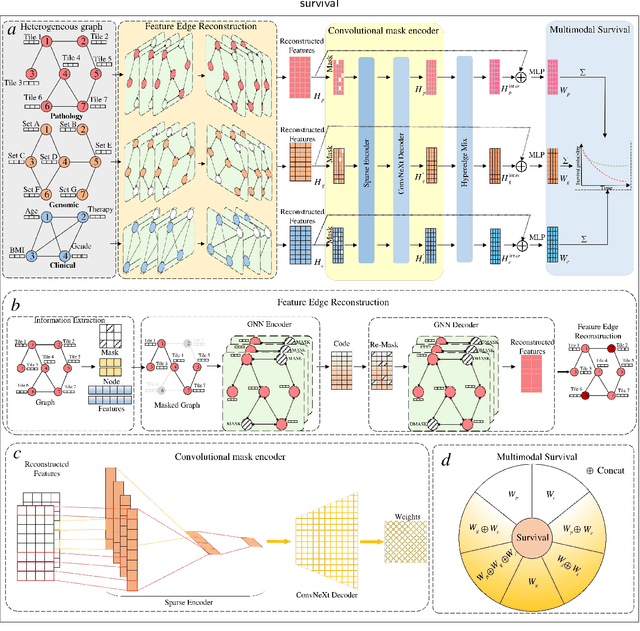
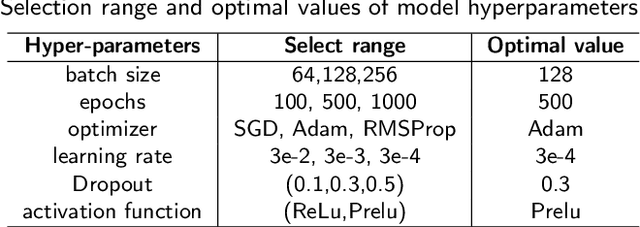
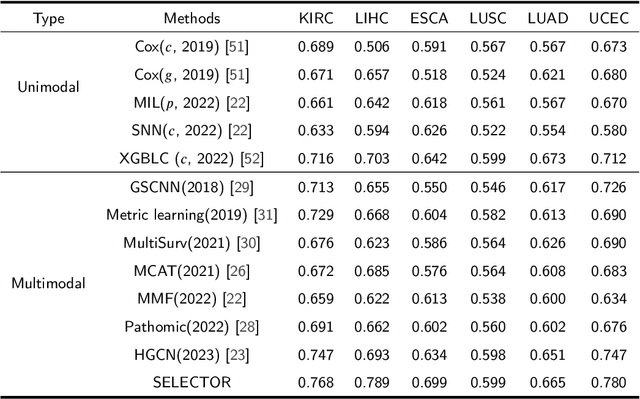
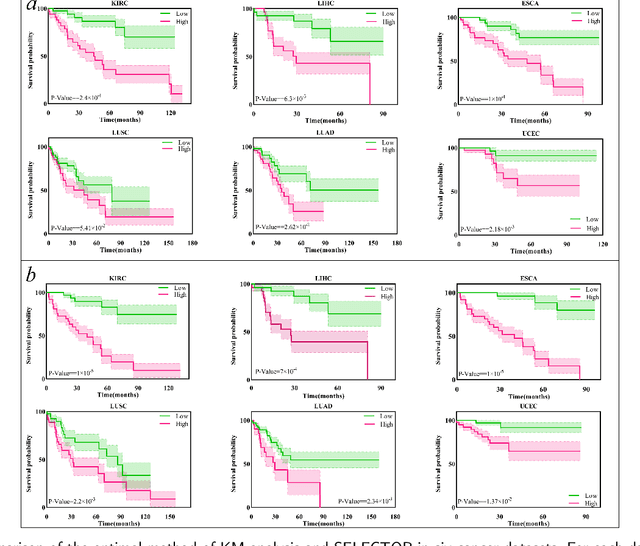
Accurately predicting the survival rate of cancer patients is crucial for aiding clinicians in planning appropriate treatment, reducing cancer-related medical expenses, and significantly enhancing patients' quality of life. Multimodal prediction of cancer patient survival offers a more comprehensive and precise approach. However, existing methods still grapple with challenges related to missing multimodal data and information interaction within modalities. This paper introduces SELECTOR, a heterogeneous graph-aware network based on convolutional mask encoders for robust multimodal prediction of cancer patient survival. SELECTOR comprises feature edge reconstruction, convolutional mask encoder, feature cross-fusion, and multimodal survival prediction modules. Initially, we construct a multimodal heterogeneous graph and employ the meta-path method for feature edge reconstruction, ensuring comprehensive incorporation of feature information from graph edges and effective embedding of nodes. To mitigate the impact of missing features within the modality on prediction accuracy, we devised a convolutional masked autoencoder (CMAE) to process the heterogeneous graph post-feature reconstruction. Subsequently, the feature cross-fusion module facilitates communication between modalities, ensuring that output features encompass all features of the modality and relevant information from other modalities. Extensive experiments and analysis on six cancer datasets from TCGA demonstrate that our method significantly outperforms state-of-the-art methods in both modality-missing and intra-modality information-confirmed cases. Our codes are made available at https://github.com/panliangrui/Selector.
ESM-NBR: fast and accurate nucleic acid-binding residue prediction via protein language model feature representation and multi-task learning
Dec 01, 2023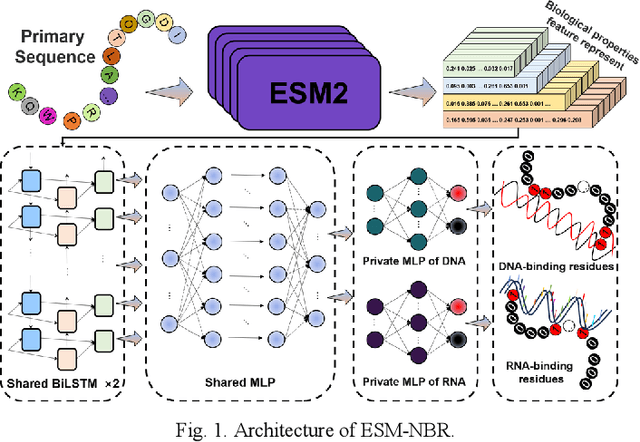
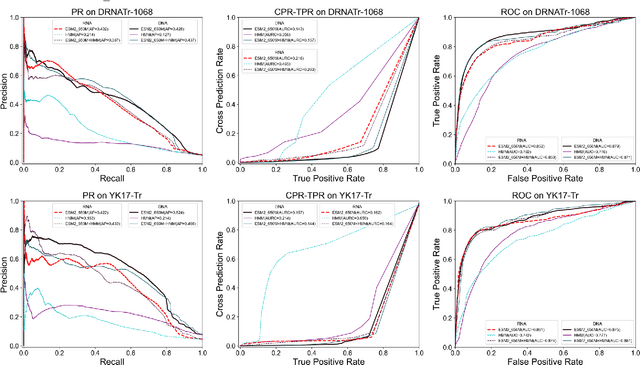
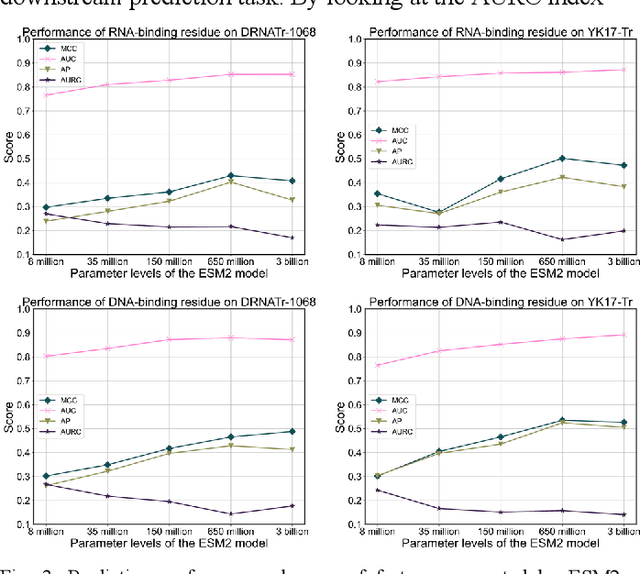
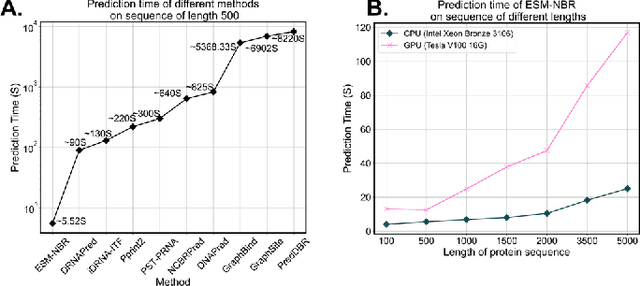
Protein-nucleic acid interactions play a very important role in a variety of biological activities. Accurate identification of nucleic acid-binding residues is a critical step in understanding the interaction mechanisms. Although many computationally based methods have been developed to predict nucleic acid-binding residues, challenges remain. In this study, a fast and accurate sequence-based method, called ESM-NBR, is proposed. In ESM-NBR, we first use the large protein language model ESM2 to extract discriminative biological properties feature representation from protein primary sequences; then, a multi-task deep learning model composed of stacked bidirectional long short-term memory (BiLSTM) and multi-layer perceptron (MLP) networks is employed to explore common and private information of DNA- and RNA-binding residues with ESM2 feature as input. Experimental results on benchmark data sets demonstrate that the prediction performance of ESM2 feature representation comprehensively outperforms evolutionary information-based hidden Markov model (HMM) features. Meanwhile, the ESM-NBR obtains the MCC values for DNA-binding residues prediction of 0.427 and 0.391 on two independent test sets, which are 18.61 and 10.45% higher than those of the second-best methods, respectively. Moreover, by completely discarding the time-cost multiple sequence alignment process, the prediction speed of ESM-NBR far exceeds that of existing methods (5.52s for a protein sequence of length 500, which is about 16 times faster than the second-fastest method). A user-friendly standalone package and the data of ESM-NBR are freely available for academic use at: https://github.com/wwzll123/ESM-NBR.
PACS: Prediction and analysis of cancer subtypes from multi-omics data based on a multi-head attention mechanism model
Aug 21, 2023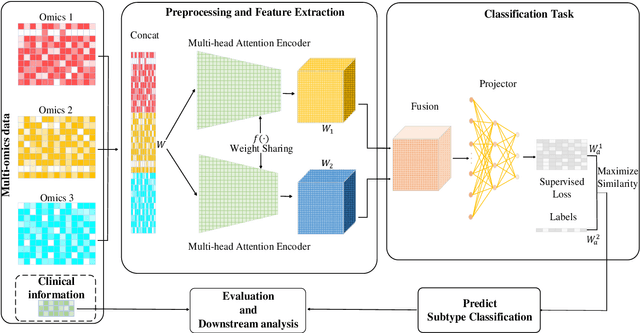
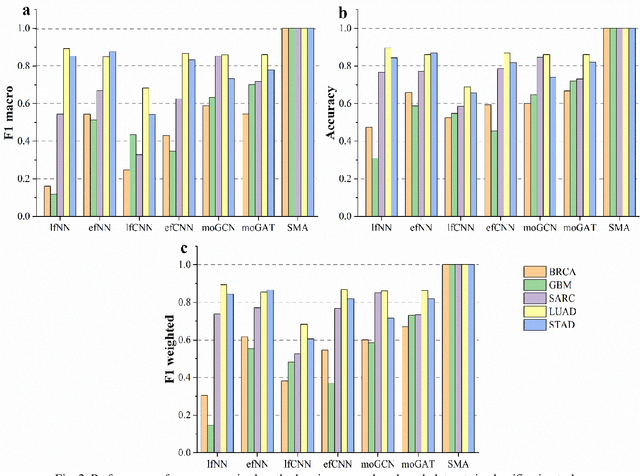
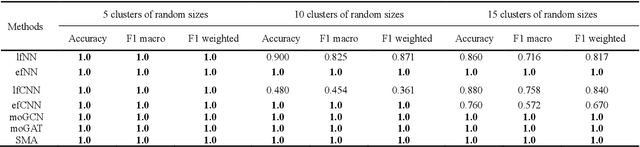
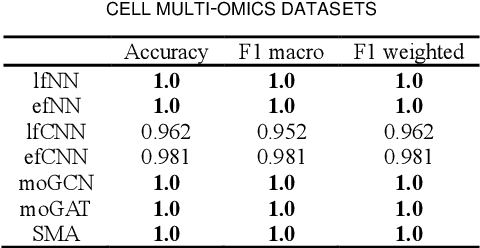
Due to the high heterogeneity and clinical characteristics of cancer, there are significant differences in multi-omic data and clinical characteristics among different cancer subtypes. Therefore, accurate classification of cancer subtypes can help doctors choose the most appropriate treatment options, improve treatment outcomes, and provide more accurate patient survival predictions. In this study, we propose a supervised multi-head attention mechanism model (SMA) to classify cancer subtypes successfully. The attention mechanism and feature sharing module of the SMA model can successfully learn the global and local feature information of multi-omics data. Second, it enriches the parameters of the model by deeply fusing multi-head attention encoders from Siamese through the fusion module. Validated by extensive experiments, the SMA model achieves the highest accuracy, F1 macroscopic, F1 weighted, and accurate classification of cancer subtypes in simulated, single-cell, and cancer multiomics datasets compared to AE, CNN, and GNN-based models. Therefore, we contribute to future research on multiomics data using our attention-based approach.
Robust Blind Source Separation by Soft Decision-Directed Non-Unitary Joint Diagonalization
Jun 28, 2021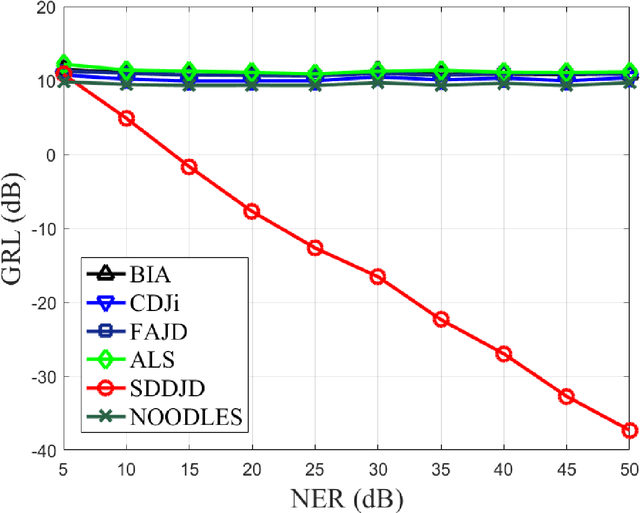
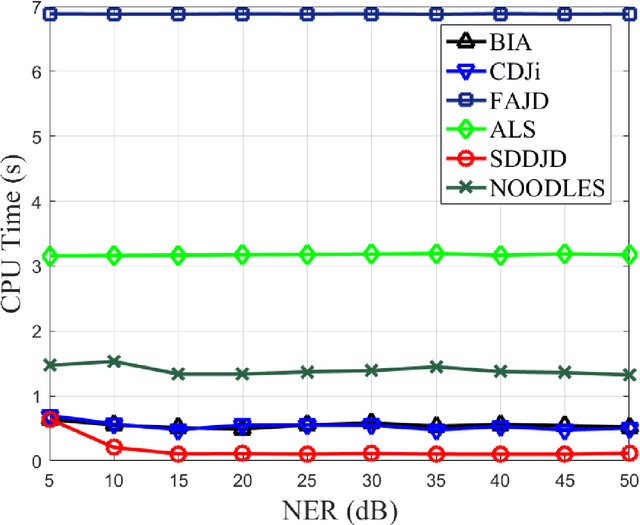
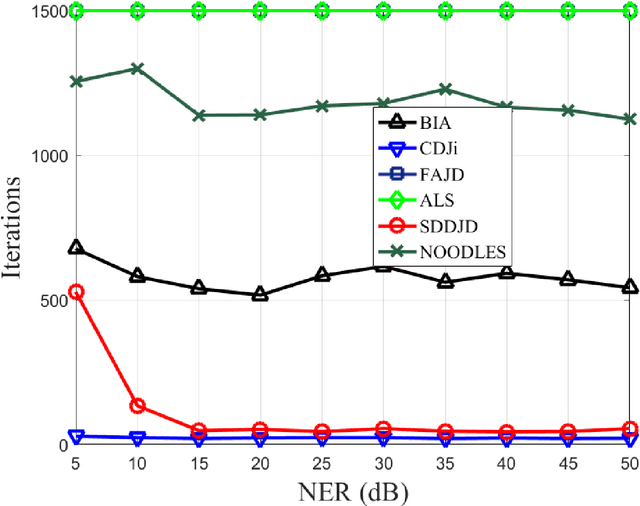
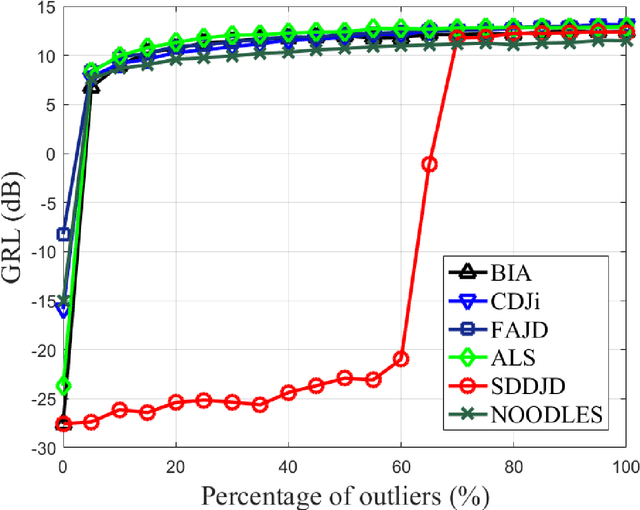
Approximate joint diagonalization of a set of matrices provides a powerful framework for numerous statistical signal processing applications. For non-unitary joint diagonalization (NUJD) based on the least-squares (LS) criterion, outliers, also referred to as anomaly or discordant observations, have a negative influence on the performance, since squaring the residuals magnifies the effects of them. To solve this problem, we propose a novel cost function that incorporates the soft decision-directed scheme into the least-squares algorithm and develops an efficient algorithm. The influence of the outliers is mitigated by applying decision-directed weights which are associated with the residual error at each iterative step. Specifically, the mixing matrix is estimated by a modified stationary point method, in which the updating direction is determined based on the linear approximation to the gradient function. Simulation results demonstrate that the proposed algorithm outperforms conventional non-unitary diagonalization algorithms in terms of both convergence performance and robustness to outliers.