 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHD-Bind: Encoding of Molecular Structure with Low Precision, Hyperdimensional Binary Representations
Mar 27, 2023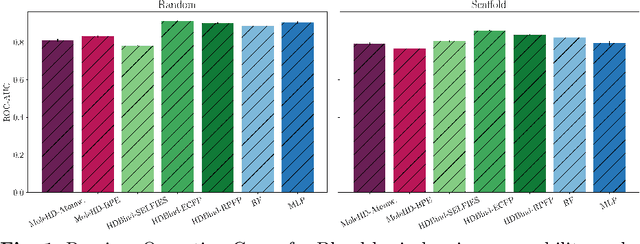
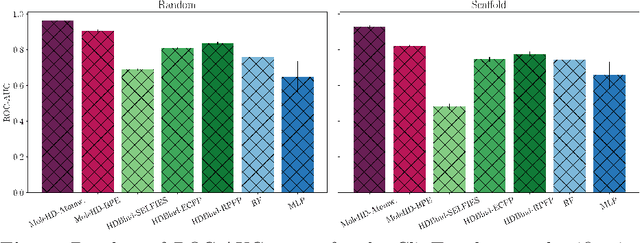
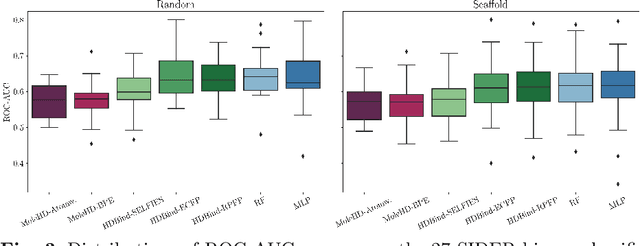
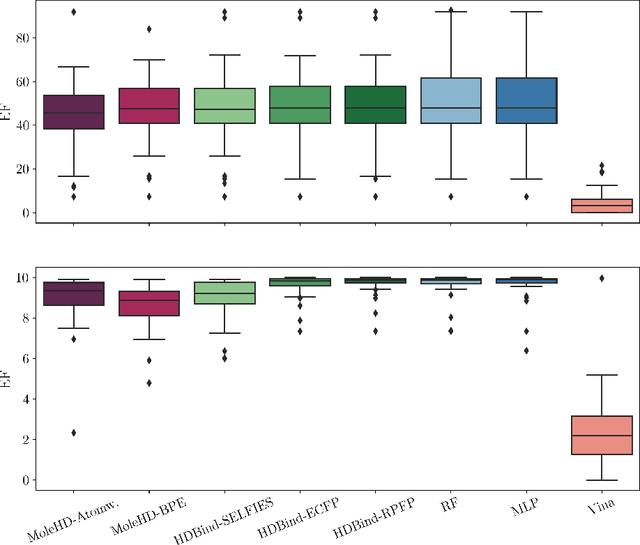
Publicly available collections of drug-like molecules have grown to comprise 10s of billions of possibilities in recent history due to advances in chemical synthesis. Traditional methods for identifying ``hit'' molecules from a large collection of potential drug-like candidates have relied on biophysical theory to compute approximations to the Gibbs free energy of the binding interaction between the drug to its protein target. A major drawback of the approaches is that they require exceptional computing capabilities to consider for even relatively small collections of molecules. Hyperdimensional Computing (HDC) is a recently proposed learning paradigm that is able to leverage low-precision binary vector arithmetic to build efficient representations of the data that can be obtained without the need for gradient-based optimization approaches that are required in many conventional machine learning and deep learning approaches. This algorithmic simplicity allows for acceleration in hardware that has been previously demonstrated for a range of application areas. We consider existing HDC approaches for molecular property classification and introduce two novel encoding algorithms that leverage the extended connectivity fingerprint (ECFP) algorithm. We show that HDC-based inference methods are as much as 90 times more efficient than more complex representative machine learning methods and achieve an acceleration of nearly 9 orders of magnitude as compared to inference with molecular docking. We demonstrate multiple approaches for the encoding of molecular data for HDC and examine their relative performance on a range of challenging molecular property prediction and drug-protein binding classification tasks. Our work thus motivates further investigation into molecular representation learning to develop ultra-efficient pre-screening tools.
Evaluating Point-Prediction Uncertainties in Neural Networks for Drug Discovery
Oct 31, 2022



Neural Network (NN) models provide potential to speed up the drug discovery process and reduce its failure rates. The success of NN models require uncertainty quantification (UQ) as drug discovery explores chemical space beyond the training data distribution. Standard NN models do not provide uncertainty information. Methods that combine Bayesian models with NN models address this issue, but are difficult to implement and more expensive to train. Some methods require changing the NN architecture or training procedure, limiting the selection of NN models. Moreover, predictive uncertainty can come from different sources. It is important to have the ability to separately model different types of predictive uncertainty, as the model can take assorted actions depending on the source of uncertainty. In this paper, we examine UQ methods that estimate different sources of predictive uncertainty for NN models aiming at drug discovery. We use our prior knowledge on chemical compounds to design the experiments. By utilizing a visualization method we create non-overlapping and chemically diverse partitions from a collection of chemical compounds. These partitions are used as training and test set splits to explore NN model uncertainty. We demonstrate how the uncertainties estimated by the selected methods describe different sources of uncertainty under different partitions and featurization schemes and the relationship to prediction error.
High-Throughput Virtual Screening of Small Molecule Inhibitors for SARS-CoV-2 Protein Targets with Deep Fusion Models
Apr 09, 2021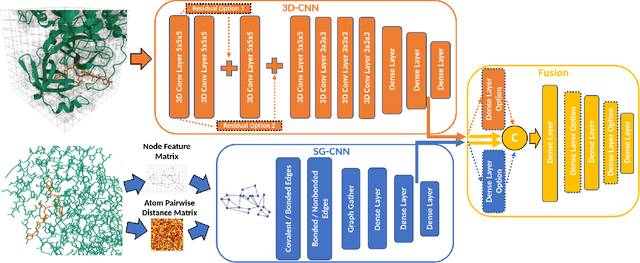
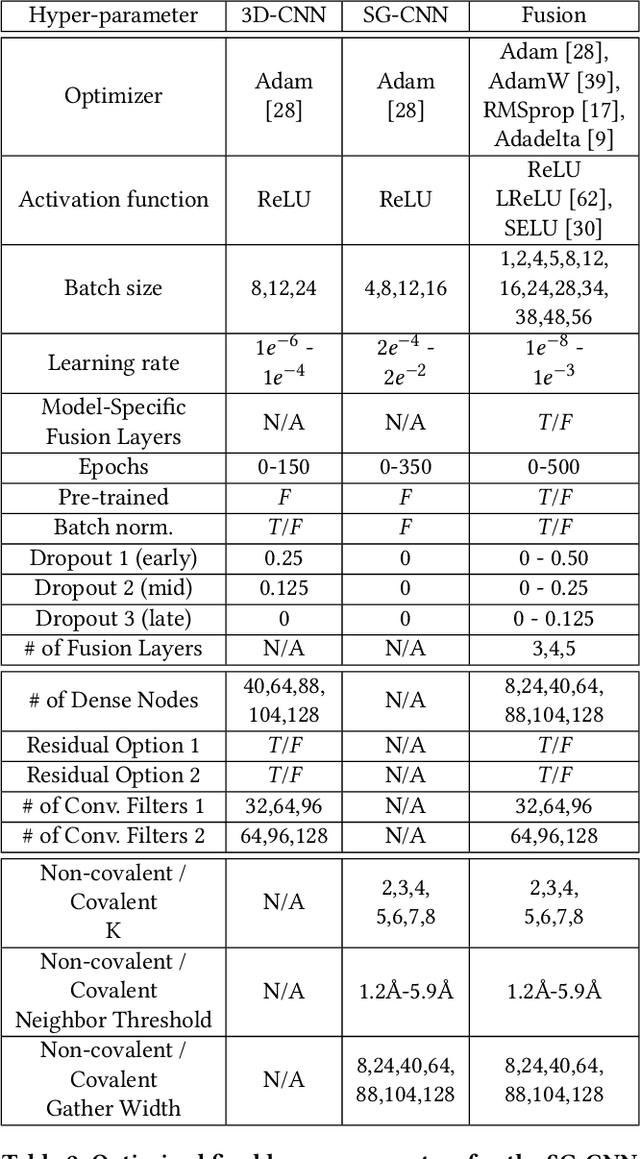
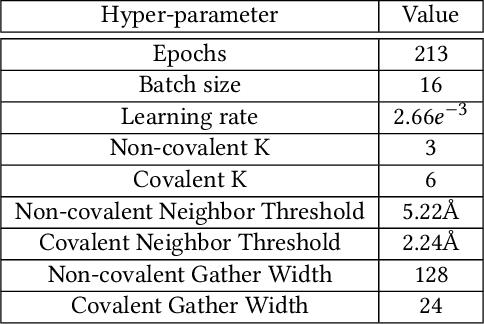
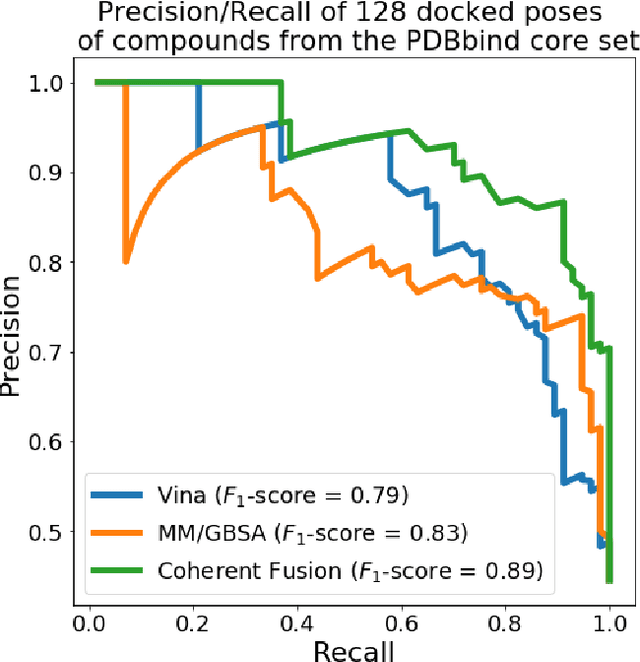
Structure-based Deep Fusion models were recently shown to outperform several physics- and machine learning-based protein-ligand binding affinity prediction methods. As part of a multi-institutional COVID-19 pandemic response, over 500 million small molecules were computationally screened against four protein structures from the novel coronavirus (SARS-CoV-2), which causes COVID-19. Three enhancements to Deep Fusion were made in order to evaluate more than 5 billion docked poses on SARS-CoV-2 protein targets. First, the Deep Fusion concept was refined by formulating the architecture as one, coherently backpropagated model (Coherent Fusion) to improve binding-affinity prediction accuracy. Secondly, the model was trained using a distributed, genetic hyper-parameter optimization. Finally, a scalable, high-throughput screening capability was developed to maximize the number of ligands evaluated and expedite the path to experimental evaluation. In this work, we present both the methods developed for machine learning-based high-throughput screening and results from using our computational pipeline to find SARS-CoV-2 inhibitors.
Improved Protein-ligand Binding Affinity Prediction with Structure-Based Deep Fusion Inference
May 17, 2020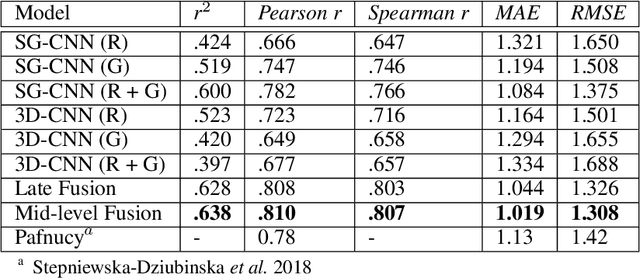
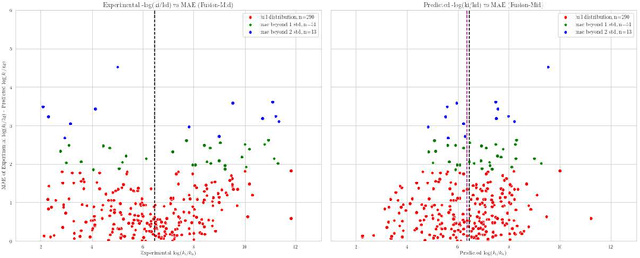
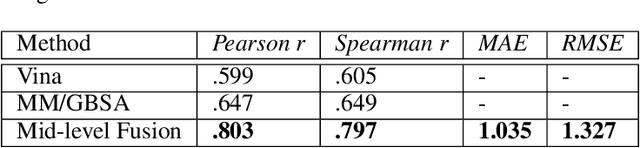
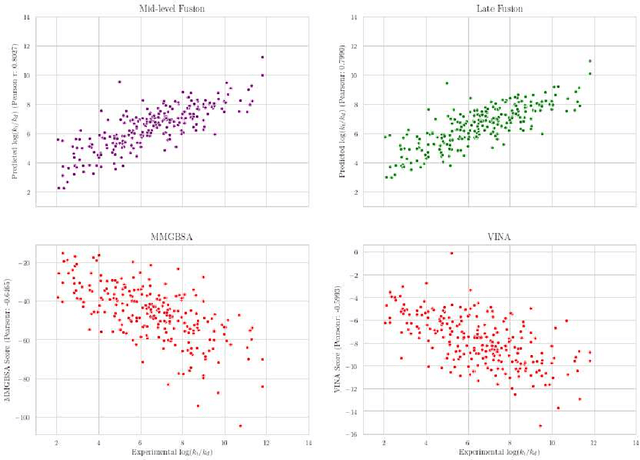
Predicting accurate protein-ligand binding affinity is important in drug discovery but remains a challenge even with computationally expensive biophysics-based energy scoring methods and state-of-the-art deep learning approaches. Despite the recent advances in the deep convolutional and graph neural network based approaches, the model performance depends on the input data representation and suffers from distinct limitations. It is natural to combine complementary features and their inference from the individual models for better predictions. We present fusion models to benefit from different feature representations of two neural network models to improve the binding affinity prediction. We demonstrate effectiveness of the proposed approach by performing experiments with the PDBBind 2016 dataset and its docking pose complexes. The results show that the proposed approach improves the overall prediction compared to the individual neural network models with greater computational efficiency than related biophysics based energy scoring functions. We also discuss the benefit of the proposed fusion inference with several example complexes. The software is made available as open source at https://github.com/llnl/fast.
AMPL: A Data-Driven Modeling Pipeline for Drug Discovery
Nov 14, 2019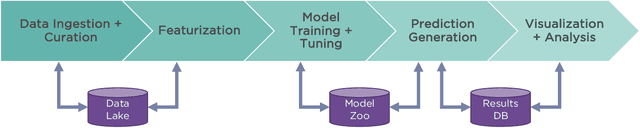

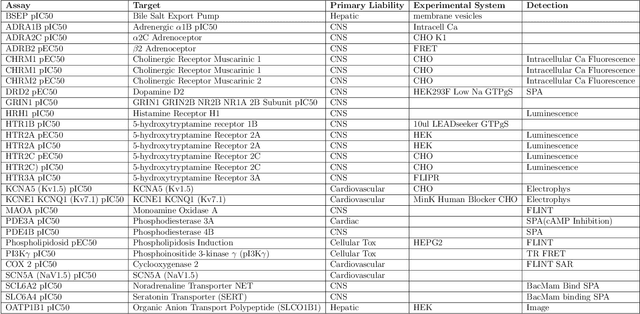
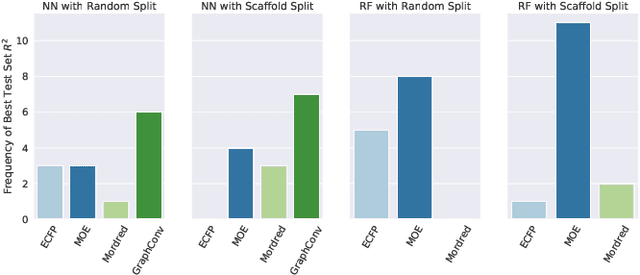
One of the key requirements for incorporating machine learning into the drug discovery process is complete reproducibility and traceability of the model building and evaluation process. With this in mind, we have developed an end-to-end modular and extensible software pipeline for building and sharing machine learning models that predict key pharma-relevant parameters. The ATOM Modeling PipeLine, or AMPL, extends the functionality of the open source library DeepChem and supports an array of machine learning and molecular featurization tools. We have benchmarked AMPL on a large collection of pharmaceutical datasets covering a wide range of parameters. As a result of these comprehensive experiments, we have found that physicochemical descriptors and deep learning-based graph representations significantly outperform traditional fingerprints in the characterization of molecular features. We have also found that dataset size is directly correlated to prediction performance, and that single-task deep learning models only outperform shallow learners if there is sufficient data. Likewise, dataset size has a direct impact on model predictivity, independent of comprehensive hyperparameter model tuning. Our findings point to the need for public dataset integration or multi-task/transfer learning approaches. Lastly, we found that uncertainty quantification (UQ) analysis may help identify model error; however, efficacy of UQ to filter predictions varies considerably between datasets and featurization/model types. AMPL is open source and available for download at http://github.com/ATOMconsortium/AMPL.
Distinguishing between Normal and Cancer Cells Using Autoencoder Node Saliency
Jan 30, 2019
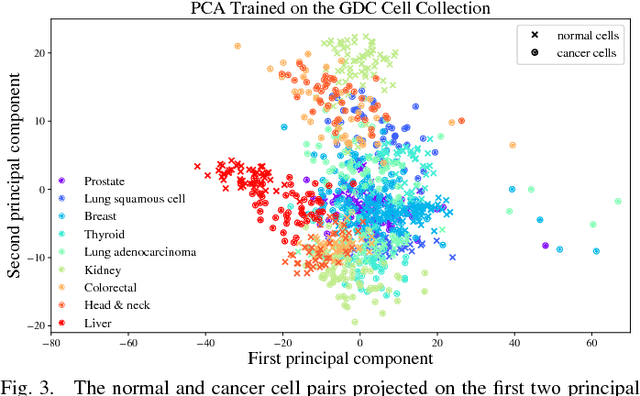
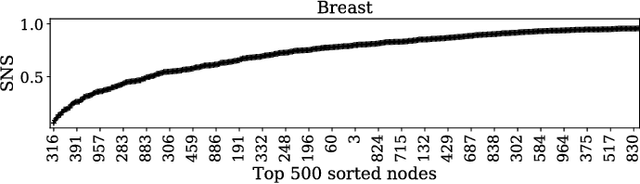
Gene expression profiles have been widely used to characterize patterns of cellular responses to diseases. As data becomes available, scalable learning toolkits become essential to processing large datasets using deep learning models to model complex biological processes. We present an autoencoder to capture nonlinear relationships recovered from gene expression profiles. The autoencoder is a nonlinear dimension reduction technique using an artificial neural network, which learns hidden representations of unlabeled data. We train the autoencoder on a large collection of tumor samples from the National Cancer Institute Genomic Data Commons, and obtain a generalized and unsupervised latent representation. We leverage a HPC-focused deep learning toolkit, Livermore Big Artificial Neural Network (LBANN) to efficiently parallelize the training algorithm, reducing computation times from several hours to a few minutes. With the trained autoencoder, we generate latent representations of a small dataset, containing pairs of normal and cancer cells of various tumor types. A novel measure called autoencoder node saliency (ANS) is introduced to identify the hidden nodes that best differentiate various pairs of cells. We compare our findings of the best classifying nodes with principal component analysis and the visualization of t-distributed stochastic neighbor embedding. We demonstrate that the autoencoder effectively extracts distinct gene features for multiple learning tasks in the dataset.