 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAMPL: A Data-Driven Modeling Pipeline for Drug Discovery
Paper and Code
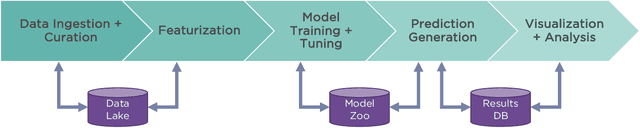

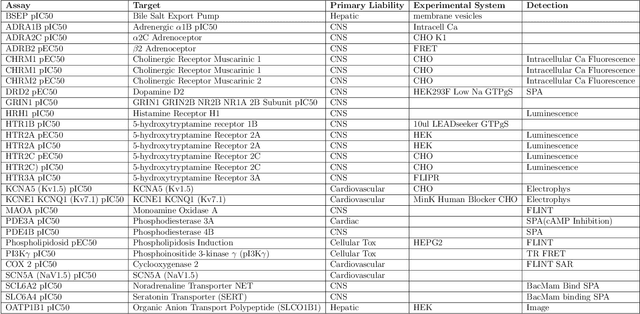
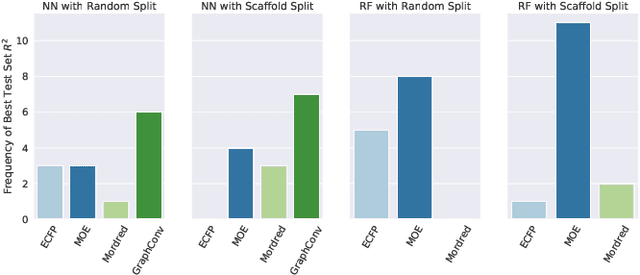
One of the key requirements for incorporating machine learning into the drug discovery process is complete reproducibility and traceability of the model building and evaluation process. With this in mind, we have developed an end-to-end modular and extensible software pipeline for building and sharing machine learning models that predict key pharma-relevant parameters. The ATOM Modeling PipeLine, or AMPL, extends the functionality of the open source library DeepChem and supports an array of machine learning and molecular featurization tools. We have benchmarked AMPL on a large collection of pharmaceutical datasets covering a wide range of parameters. As a result of these comprehensive experiments, we have found that physicochemical descriptors and deep learning-based graph representations significantly outperform traditional fingerprints in the characterization of molecular features. We have also found that dataset size is directly correlated to prediction performance, and that single-task deep learning models only outperform shallow learners if there is sufficient data. Likewise, dataset size has a direct impact on model predictivity, independent of comprehensive hyperparameter model tuning. Our findings point to the need for public dataset integration or multi-task/transfer learning approaches. Lastly, we found that uncertainty quantification (UQ) analysis may help identify model error; however, efficacy of UQ to filter predictions varies considerably between datasets and featurization/model types. AMPL is open source and available for download at http://github.com/ATOMconsortium/AMPL.