 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEvaluating Point-Prediction Uncertainties in Neural Networks for Drug Discovery
Oct 31, 2022



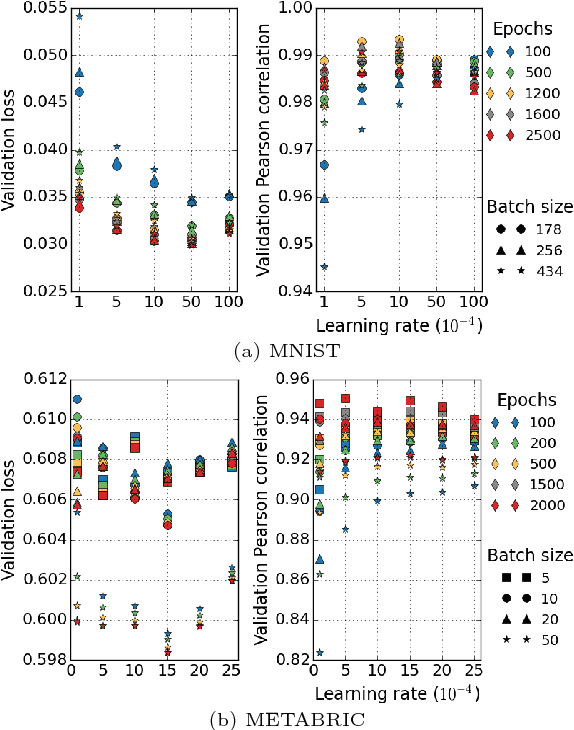
Neural Network (NN) models provide potential to speed up the drug discovery process and reduce its failure rates. The success of NN models require uncertainty quantification (UQ) as drug discovery explores chemical space beyond the training data distribution. Standard NN models do not provide uncertainty information. Methods that combine Bayesian models with NN models address this issue, but are difficult to implement and more expensive to train. Some methods require changing the NN architecture or training procedure, limiting the selection of NN models. Moreover, predictive uncertainty can come from different sources. It is important to have the ability to separately model different types of predictive uncertainty, as the model can take assorted actions depending on the source of uncertainty. In this paper, we examine UQ methods that estimate different sources of predictive uncertainty for NN models aiming at drug discovery. We use our prior knowledge on chemical compounds to design the experiments. By utilizing a visualization method we create non-overlapping and chemically diverse partitions from a collection of chemical compounds. These partitions are used as training and test set splits to explore NN model uncertainty. We demonstrate how the uncertainties estimated by the selected methods describe different sources of uncertainty under different partitions and featurization schemes and the relationship to prediction error.
Distinguishing between Normal and Cancer Cells Using Autoencoder Node Saliency
Jan 30, 2019
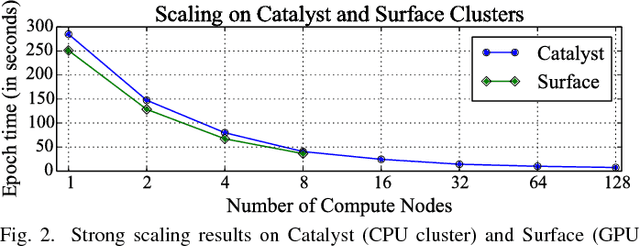
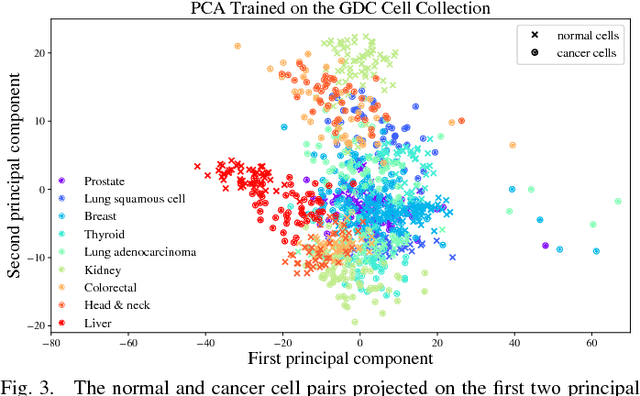
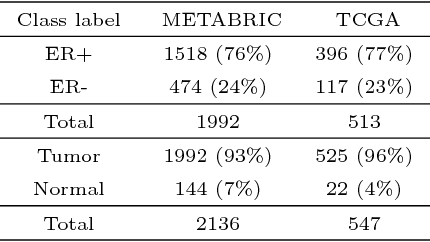
Gene expression profiles have been widely used to characterize patterns of cellular responses to diseases. As data becomes available, scalable learning toolkits become essential to processing large datasets using deep learning models to model complex biological processes. We present an autoencoder to capture nonlinear relationships recovered from gene expression profiles. The autoencoder is a nonlinear dimension reduction technique using an artificial neural network, which learns hidden representations of unlabeled data. We train the autoencoder on a large collection of tumor samples from the National Cancer Institute Genomic Data Commons, and obtain a generalized and unsupervised latent representation. We leverage a HPC-focused deep learning toolkit, Livermore Big Artificial Neural Network (LBANN) to efficiently parallelize the training algorithm, reducing computation times from several hours to a few minutes. With the trained autoencoder, we generate latent representations of a small dataset, containing pairs of normal and cancer cells of various tumor types. A novel measure called autoencoder node saliency (ANS) is introduced to identify the hidden nodes that best differentiate various pairs of cells. We compare our findings of the best classifying nodes with principal component analysis and the visualization of t-distributed stochastic neighbor embedding. We demonstrate that the autoencoder effectively extracts distinct gene features for multiple learning tasks in the dataset.
Autoencoder Node Saliency: Selecting Relevant Latent Representations
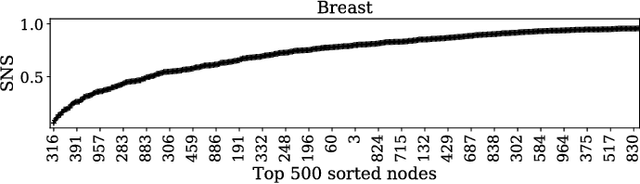
Mar 08, 2018
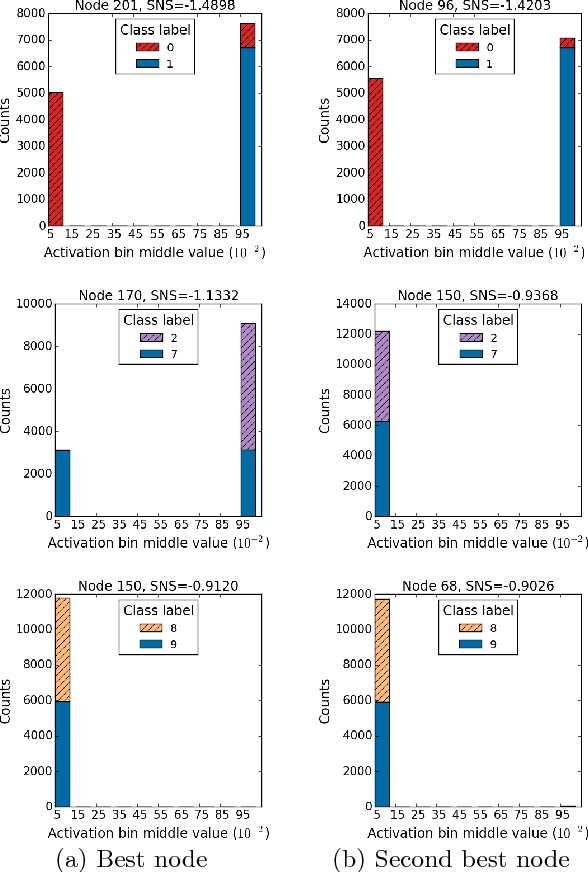
The autoencoder is an artificial neural network model that learns hidden representations of unlabeled data. With a linear transfer function it is similar to the principal component analysis (PCA). While both methods use weight vectors for linear transformations, the autoencoder does not come with any indication similar to the eigenvalues in PCA that are paired with the eigenvectors. We propose a novel supervised node saliency (SNS) method that ranks the hidden nodes by comparing class distributions of latent representations against a fixed reference distribution. The latent representations of a hidden node can be described using a one-dimensional histogram. We apply normalized entropy difference (NED) to measure the "interestingness" of the histograms, and conclude a property for NED values to identify a good classifying node. By applying our methods to real data sets, we demonstrate the ability of SNS to explain what the trained autoencoders have learned.