 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEvaluating Point-Prediction Uncertainties in Neural Networks for Drug Discovery
Oct 31, 2022



Neural Network (NN) models provide potential to speed up the drug discovery process and reduce its failure rates. The success of NN models require uncertainty quantification (UQ) as drug discovery explores chemical space beyond the training data distribution. Standard NN models do not provide uncertainty information. Methods that combine Bayesian models with NN models address this issue, but are difficult to implement and more expensive to train. Some methods require changing the NN architecture or training procedure, limiting the selection of NN models. Moreover, predictive uncertainty can come from different sources. It is important to have the ability to separately model different types of predictive uncertainty, as the model can take assorted actions depending on the source of uncertainty. In this paper, we examine UQ methods that estimate different sources of predictive uncertainty for NN models aiming at drug discovery. We use our prior knowledge on chemical compounds to design the experiments. By utilizing a visualization method we create non-overlapping and chemically diverse partitions from a collection of chemical compounds. These partitions are used as training and test set splits to explore NN model uncertainty. We demonstrate how the uncertainties estimated by the selected methods describe different sources of uncertainty under different partitions and featurization schemes and the relationship to prediction error.
High-Throughput Virtual Screening of Small Molecule Inhibitors for SARS-CoV-2 Protein Targets with Deep Fusion Models
Apr 09, 2021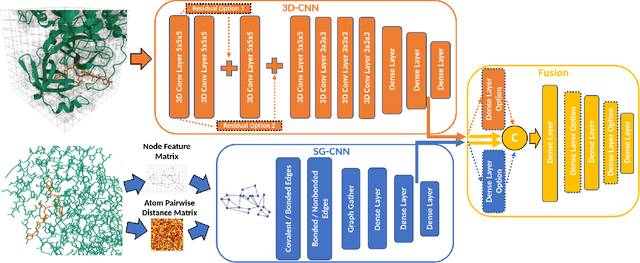
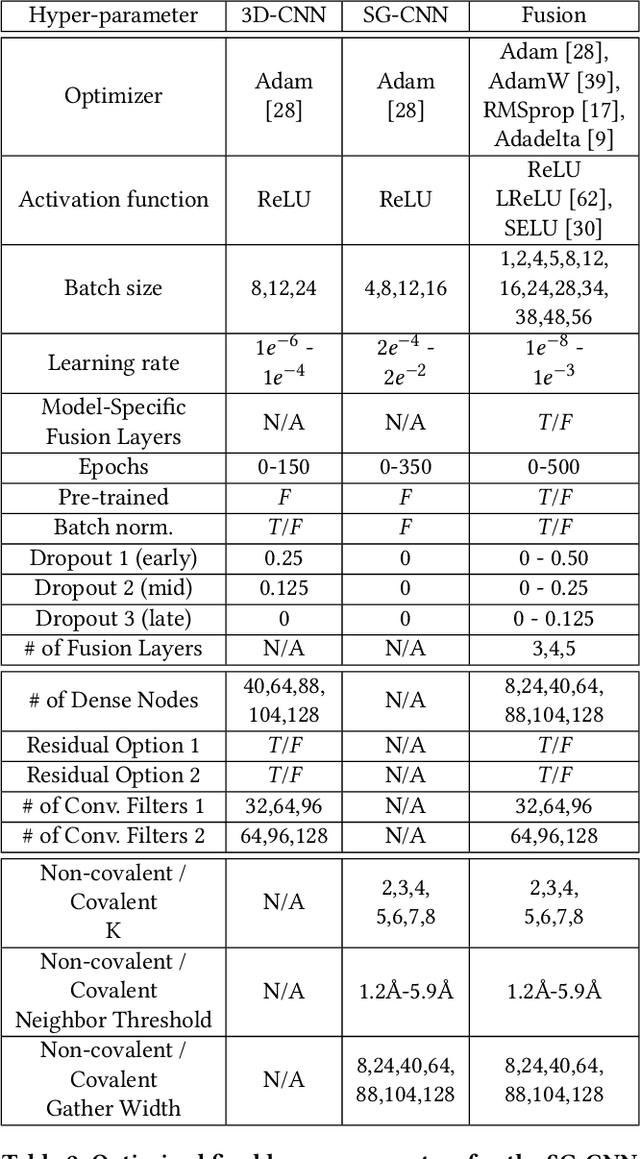
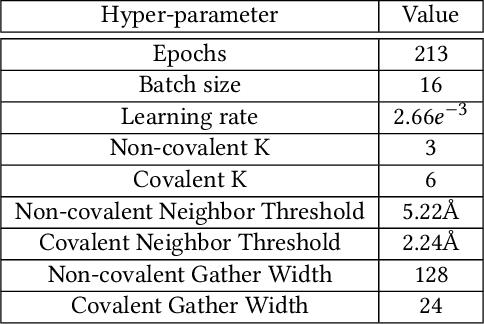
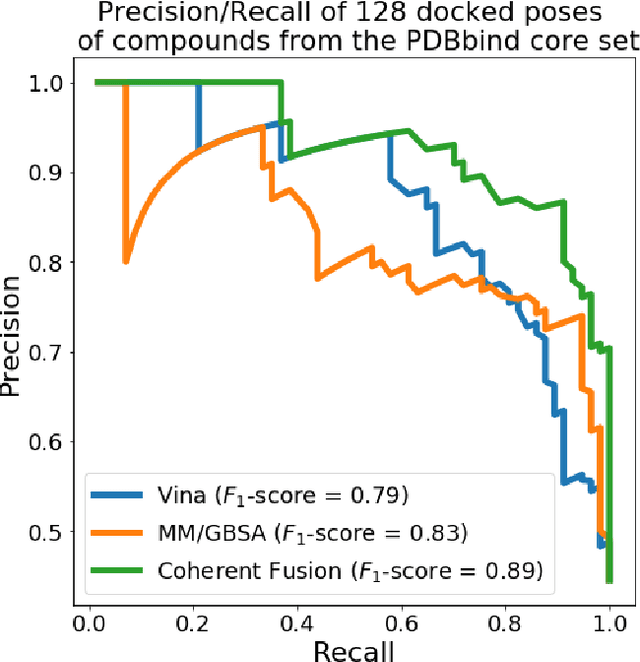
Structure-based Deep Fusion models were recently shown to outperform several physics- and machine learning-based protein-ligand binding affinity prediction methods. As part of a multi-institutional COVID-19 pandemic response, over 500 million small molecules were computationally screened against four protein structures from the novel coronavirus (SARS-CoV-2), which causes COVID-19. Three enhancements to Deep Fusion were made in order to evaluate more than 5 billion docked poses on SARS-CoV-2 protein targets. First, the Deep Fusion concept was refined by formulating the architecture as one, coherently backpropagated model (Coherent Fusion) to improve binding-affinity prediction accuracy. Secondly, the model was trained using a distributed, genetic hyper-parameter optimization. Finally, a scalable, high-throughput screening capability was developed to maximize the number of ligands evaluated and expedite the path to experimental evaluation. In this work, we present both the methods developed for machine learning-based high-throughput screening and results from using our computational pipeline to find SARS-CoV-2 inhibitors.