 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSpecHD: Hyperdimensional Computing Framework for FPGA-based Mass Spectrometry Clustering
Nov 20, 2023Mass spectrometry-based proteomics is a key enabler for personalized healthcare, providing a deep dive into the complex protein compositions of biological systems. This technology has vast applications in biotechnology and biomedicine but faces significant computational bottlenecks. Current methodologies often require multiple hours or even days to process extensive datasets, particularly in the domain of spectral clustering. To tackle these inefficiencies, we introduce SpecHD, a hyperdimensional computing (HDC) framework supplemented by an FPGA-accelerated architecture with integrated near-storage preprocessing. Utilizing streamlined binary operations in an HDC environment, SpecHD capitalizes on the low-latency and parallel capabilities of FPGAs. This approach markedly improves clustering speed and efficiency, serving as a catalyst for real-time, high-throughput data analysis in future healthcare applications. Our evaluations demonstrate that SpecHD not only maintains but often surpasses existing clustering quality metrics while drastically cutting computational time. Specifically, it can cluster a large-scale human proteome dataset-comprising 25 million MS/MS spectra and 131 GB of MS data-in just 5 minutes. With energy efficiency exceeding 31x and a speedup factor that spans a range of 6x to 54x over existing state of-the-art solutions, SpecHD emerges as a promising solution for the rapid analysis of mass spectrometry data with great implications for personalized healthcare.
HD-Bind: Encoding of Molecular Structure with Low Precision, Hyperdimensional Binary Representations
Mar 27, 2023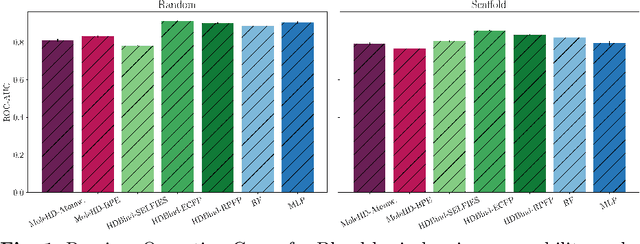
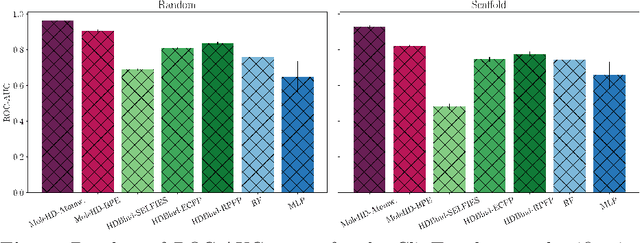
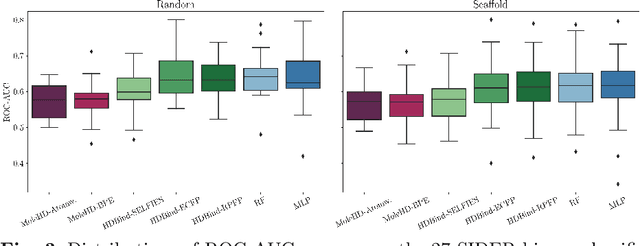
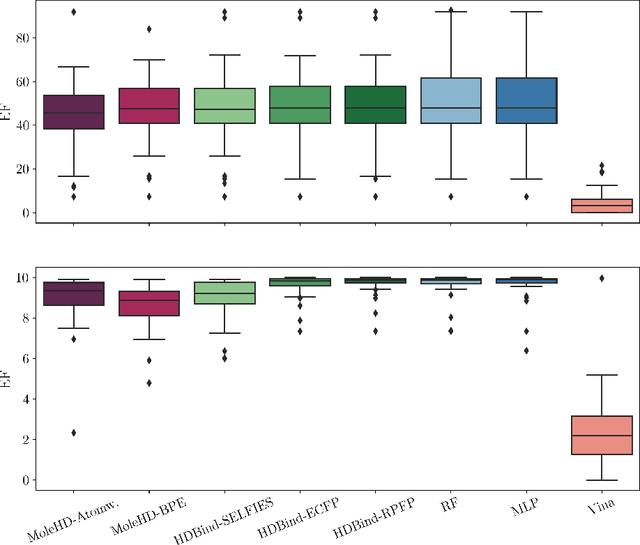
Publicly available collections of drug-like molecules have grown to comprise 10s of billions of possibilities in recent history due to advances in chemical synthesis. Traditional methods for identifying ``hit'' molecules from a large collection of potential drug-like candidates have relied on biophysical theory to compute approximations to the Gibbs free energy of the binding interaction between the drug to its protein target. A major drawback of the approaches is that they require exceptional computing capabilities to consider for even relatively small collections of molecules. Hyperdimensional Computing (HDC) is a recently proposed learning paradigm that is able to leverage low-precision binary vector arithmetic to build efficient representations of the data that can be obtained without the need for gradient-based optimization approaches that are required in many conventional machine learning and deep learning approaches. This algorithmic simplicity allows for acceleration in hardware that has been previously demonstrated for a range of application areas. We consider existing HDC approaches for molecular property classification and introduce two novel encoding algorithms that leverage the extended connectivity fingerprint (ECFP) algorithm. We show that HDC-based inference methods are as much as 90 times more efficient than more complex representative machine learning methods and achieve an acceleration of nearly 9 orders of magnitude as compared to inference with molecular docking. We demonstrate multiple approaches for the encoding of molecular data for HDC and examine their relative performance on a range of challenging molecular property prediction and drug-protein binding classification tasks. Our work thus motivates further investigation into molecular representation learning to develop ultra-efficient pre-screening tools.