 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOmniCellTOSG: The First Cell Text-Omic Signaling Graphs Dataset for Joint LLM and GNN Modeling
Apr 02, 2025Complex cell signaling systems -- governed by varying protein abundances and interactions -- generate diverse cell types across organs. These systems evolve under influences such as age, sex, diet, environmental exposures, and diseases, making them challenging to decode given the involvement of tens of thousands of genes and proteins. Recently, hundreds of millions of single-cell omics data have provided a robust foundation for understanding these signaling networks within various cell subpopulations and conditions. Inspired by the success of large foundation models (for example, large language models and large vision models) pre-trained on massive datasets, we introduce OmniCellTOSG, the first dataset of cell text-omic signaling graphs (TOSGs). Each TOSG represents the signaling network of an individual or meta-cell and is labeled with information such as organ, disease, sex, age, and cell subtype. OmniCellTOSG offers two key contributions. First, it introduces a novel graph model that integrates human-readable annotations -- such as biological functions, cellular locations, signaling pathways, related diseases, and drugs -- with quantitative gene and protein abundance data, enabling graph reasoning to decode cell signaling. This approach calls for new joint models combining large language models and graph neural networks. Second, the dataset is built from single-cell RNA sequencing data of approximately 120 million cells from diverse tissues and conditions (healthy and diseased) and is fully compatible with PyTorch. This facilitates the development of innovative cell signaling models that could transform research in life sciences, healthcare, and precision medicine. The OmniCellTOSG dataset is continuously expanding and will be updated regularly. The dataset and code are available at https://github.com/FuhaiLiAiLab/OmniCellTOSG.
KoGNER: A Novel Framework for Knowledge Graph Distillation on Biomedical Named Entity Recognition
Mar 19, 2025Named Entity Recognition (NER) is a fundamental task in Natural Language Processing (NLP) that plays a crucial role in information extraction, question answering, and knowledge-based systems. Traditional deep learning-based NER models often struggle with domain-specific generalization and suffer from data sparsity issues. In this work, we introduce Knowledge Graph distilled for Named Entity Recognition (KoGNER), a novel approach that integrates Knowledge Graph (KG) distillation into NER models to enhance entity recognition performance. Our framework leverages structured knowledge representations from KGs to enrich contextual embeddings, thereby improving entity classification and reducing ambiguity in entity detection. KoGNER employs a two-step process: (1) Knowledge Distillation, where external knowledge sources are distilled into a lightweight representation for seamless integration with NER models, and (2) Entity-Aware Augmentation, which integrates contextual embeddings that have been enriched with knowledge graph information directly into GNN, thereby improving the model's ability to understand and represent entity relationships. Experimental results on benchmark datasets demonstrate that KoGNER achieves state-of-the-art performance, outperforming finetuned NER models and LLMs by a significant margin. These findings suggest that leveraging knowledge graphs as auxiliary information can significantly improve NER accuracy, making KoGNER a promising direction for future research in knowledge-aware NLP.
GraphSeqLM: A Unified Graph Language Framework for Omic Graph Learning
Dec 20, 2024

The integration of multi-omic data is pivotal for understanding complex diseases, but its high dimensionality and noise present significant challenges. Graph Neural Networks (GNNs) offer a robust framework for analyzing large-scale signaling pathways and protein-protein interaction networks, yet they face limitations in expressivity when capturing intricate biological relationships. To address this, we propose Graph Sequence Language Model (GraphSeqLM), a framework that enhances GNNs with biological sequence embeddings generated by Large Language Models (LLMs). These embeddings encode structural and biological properties of DNA, RNA, and proteins, augmenting GNNs with enriched features for analyzing sample-specific multi-omic data. By integrating topological, sequence-derived, and biological information, GraphSeqLM demonstrates superior predictive accuracy and outperforms existing methods, paving the way for more effective multi-omic data integration in precision medicine.
Highly Accurate Disease Diagnosis and Highly Reproducible Biomarker Identification with PathFormer
Feb 11, 2024



Biomarker identification is critical for precise disease diagnosis and understanding disease pathogenesis in omics data analysis, like using fold change and regression analysis. Graph neural networks (GNNs) have been the dominant deep learning model for analyzing graph-structured data. However, we found two major limitations of existing GNNs in omics data analysis, i.e., limited-prediction (diagnosis) accuracy and limited-reproducible biomarker identification capacity across multiple datasets. The root of the challenges is the unique graph structure of biological signaling pathways, which consists of a large number of targets and intensive and complex signaling interactions among these targets. To resolve these two challenges, in this study, we presented a novel GNN model architecture, named PathFormer, which systematically integrate signaling network, priori knowledge and omics data to rank biomarkers and predict disease diagnosis. In the comparison results, PathFormer outperformed existing GNN models significantly in terms of highly accurate prediction capability ( 30% accuracy improvement in disease diagnosis compared with existing GNN models) and high reproducibility of biomarker ranking across different datasets. The improvement was confirmed using two independent Alzheimer's Disease (AD) and cancer transcriptomic datasets. The PathFormer model can be directly applied to other omics data analysis studies.
Universal Normalization Enhanced Graph Representation Learning for Gene Network Prediction
Sep 01, 2023



Effective gene network representation learning is of great importance in bioinformatics to predict/understand the relation of gene profiles and disease phenotypes. Though graph neural networks (GNNs) have been the dominant architecture for analyzing various graph-structured data like social networks, their predicting on gene networks often exhibits subpar performance. In this paper, we formally investigate the gene network representation learning problem and characterize a notion of \textit{universal graph normalization}, where graph normalization can be applied in an universal manner to maximize the expressive power of GNNs while maintaining the stability. We propose a novel UNGNN (Universal Normalized GNN) framework, which leverages universal graph normalization in both the message passing phase and readout layer to enhance the performance of a base GNN. UNGNN has a plug-and-play property and can be combined with any GNN backbone in practice. A comprehensive set of experiments on gene-network-based bioinformatical tasks demonstrates that our UNGNN model significantly outperforms popular GNN benchmarks and provides an overall performance improvement of 16 $\%$ on average compared to previous state-of-the-art (SOTA) baselines. Furthermore, we also evaluate our theoretical findings on other graph datasets where the universal graph normalization is solvable, and we observe that UNGNN consistently achieves the superior performance.
Towards Arbitrarily Expressive GNNs in $O(n^2)$ Space by Rethinking Folklore Weisfeiler-Lehman
Jun 05, 2023Message passing neural networks (MPNNs) have emerged as the most popular framework of graph neural networks (GNNs) in recent years. However, their expressive power is limited by the 1-dimensional Weisfeiler-Lehman (1-WL) test. Some works are inspired by $k$-WL/FWL (Folklore WL) and design the corresponding neural versions. Despite the high expressive power, there are serious limitations in this line of research. In particular, (1) $k$-WL/FWL requires at least $O(n^k)$ space complexity, which is impractical for large graphs even when $k=3$; (2) The design space of $k$-WL/FWL is rigid, with the only adjustable hyper-parameter being $k$. To tackle the first limitation, we propose an extension, $(k, t)$-FWL. We theoretically prove that even if we fix the space complexity to $O(n^2)$ in $(k, t)$-FWL, we can construct an expressiveness hierarchy up to solving the graph isomorphism problem. To tackle the second problem, we propose $k$-FWL+, which considers any equivariant set as neighbors instead of all nodes, thereby greatly expanding the design space of $k$-FWL. Combining these two modifications results in a flexible and powerful framework $(k, t)$-FWL+. We demonstrate $(k, t)$-FWL+ can implement most existing models with matching expressiveness. We then introduce an instance of $(k,t)$-FWL+ called Neighborhood$^2$-FWL (N$^2$-FWL), which is practically and theoretically sound. We prove that N$^2$-FWL is no less powerful than 3-WL, can encode many substructures while only requiring $O(n^2)$ space. Finally, we design its neural version named N$^2$-GNN and evaluate its performance on various tasks. N$^2$-GNN achieves superior performance on almost all tasks, with record-breaking results on ZINC-Subset (0.059) and ZINC-Full (0.013), outperforming previous state-of-the-art results by 10.6% and 40.9%, respectively.
Interpreting mechanism of Synergism of drug combinations using attention based hierarchical graph pooling
Sep 19, 2022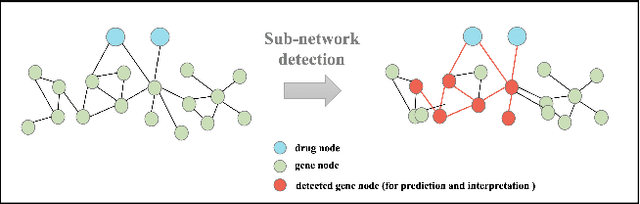
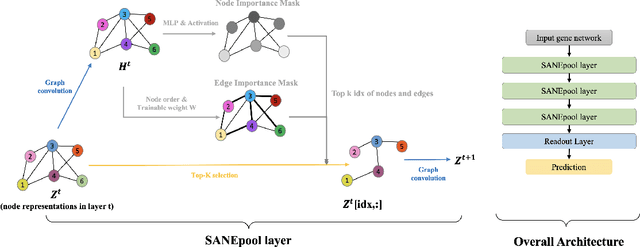
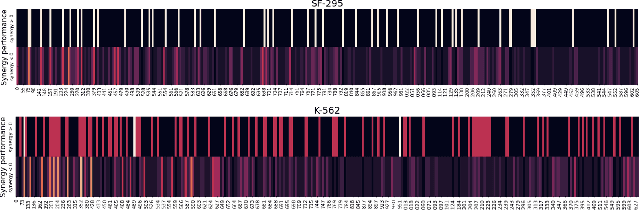
The synergistic drug combinations provide huge potentials to enhance therapeutic efficacy and to reduce adverse reactions. However, effective and synergistic drug combination prediction remains an open question because of the unknown causal disease signaling pathways. Though various deep learning (AI) models have been proposed to quantitatively predict the synergism of drug combinations. The major limitation of existing deep learning methods is that they are inherently not interpretable, which makes the conclusion of AI models un-transparent to human experts, henceforth limiting the robustness of the model conclusion and the implementation ability of these models in the real-world human-AI healthcare. In this paper, we develop an interpretable graph neural network (GNN) that reveals the underlying essential therapeutic targets and mechanism of the synergy (MoS) by mining the sub-molecular network of great importance. The key point of the interpretable GNN prediction model is a novel graph pooling layer, Self-Attention based Node and Edge pool (henceforth SANEpool), that can compute the attention score (importance) of nodes and edges based on the node features and the graph topology. As such, the proposed GNN model provides a systematic way to predict and interpret the drug combination synergism based on the detected crucial sub-molecular network. We evaluate SANEpool on molecular networks formulated by genes from 46 core cancer signaling pathways and drug combinations from NCI ALMANAC drug combination screening data. The experimental results indicate that 1) SANEpool can achieve the current state-of-art performance among other popular graph neural networks; and 2) the sub-molecular network detected by SANEpool are self-explainable and salient for identifying synergistic drug combinations.
How Powerful are K-hop Message Passing Graph Neural Networks
May 26, 2022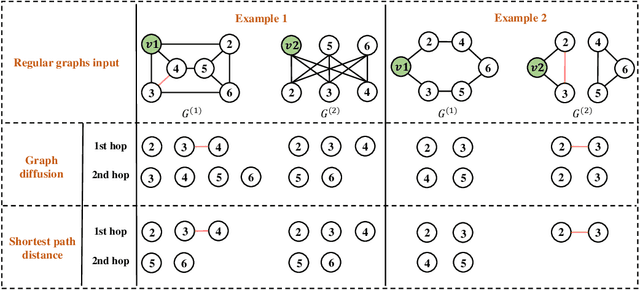
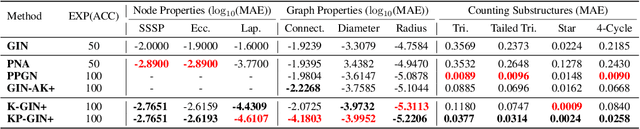
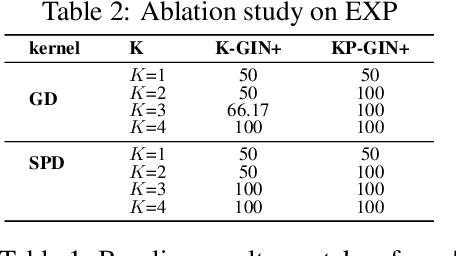
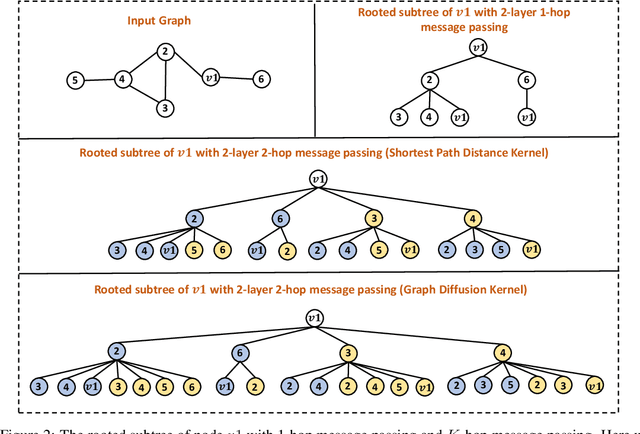
The most popular design paradigm for Graph Neural Networks (GNNs) is 1-hop message passing -- aggregating features from 1-hop neighbors repeatedly. However, the expressive power of 1-hop message passing is bounded by the Weisfeiler-Lehman (1-WL) test. Recently, researchers extended 1-hop message passing to K-hop message passing by aggregating information from K-hop neighbors of nodes simultaneously. However, there is no work on analyzing the expressive power of K-hop message passing. In this work, we theoretically characterize the expressive power of K-hop message passing. Specifically, we first formally differentiate two kinds of kernels of K-hop message passing which are often misused in previous works. We then characterize the expressive power of K-hop message passing by showing that it is more powerful than 1-hop message passing. Despite the higher expressive power, we show that K-hop message passing still cannot distinguish some simple regular graphs. To further enhance its expressive power, we introduce a KP-GNN framework, which improves K-hop message passing by leveraging the peripheral subgraph information in each hop. We prove that KP-GNN can distinguish almost all regular graphs including some distance regular graphs which could not be distinguished by previous distance encoding methods. Experimental results verify the expressive power and effectiveness of KP-GNN. KP-GNN achieves competitive results across all benchmark datasets.
PACE: A Parallelizable Computation Encoder for Directed Acyclic Graphs
Mar 19, 2022
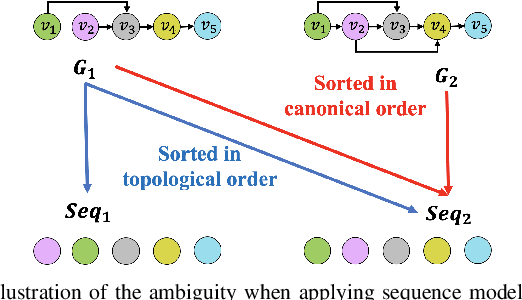
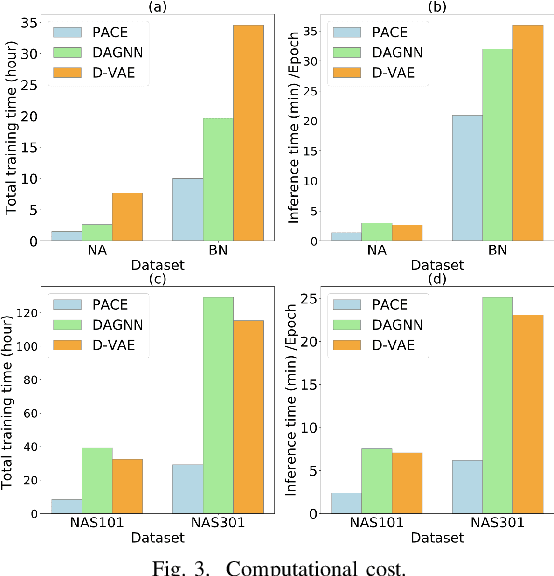

Optimization of directed acyclic graph (DAG) structures has many applications, such as neural architecture search (NAS) and probabilistic graphical model learning. Encoding DAGs into real vectors is a dominant component in most neural-network-based DAG optimization frameworks. Currently, most DAG encoders use an asynchronous message passing scheme which sequentially processes nodes according to the dependency between nodes in a DAG. That is, a node must not be processed until all its predecessors are processed. As a result, they are inherently not parallelizable. In this work, we propose a Parallelizable Attention-based Computation structure Encoder (PACE) that processes nodes simultaneously and encodes DAGs in parallel. We demonstrate the superiority of PACE through encoder-dependent optimization subroutines that search the optimal DAG structure based on the learned DAG embeddings. Experiments show that PACE not only improves the effectiveness over previous sequential DAG encoders with a significantly boosted training and inference speed, but also generates smooth latent (DAG encoding) spaces that are beneficial to downstream optimization subroutines. Our source code is available at \url{https://github.com/zehaodong/PACE}
Interpretable Drug Synergy Prediction with Graph Neural Networks for Human-AI Collaboration in Healthcare
May 14, 2021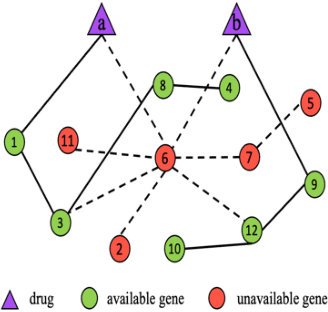
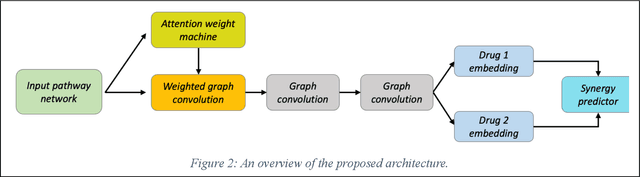
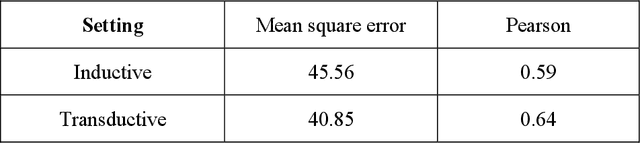
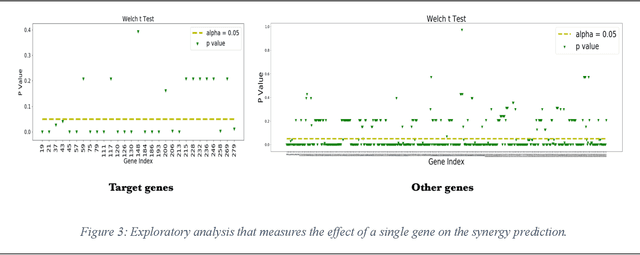
We investigate molecular mechanisms of resistant or sensitive response of cancer drug combination therapies in an inductive and interpretable manner. Though deep learning algorithms are widely used in the drug synergy prediction problem, it is still an open problem to formulate the prediction model with biological meaning to investigate the mysterious mechanisms of synergy (MoS) for the human-AI collaboration in healthcare systems. To address the challenges, we propose a deep graph neural network, IDSP (Interpretable Deep Signaling Pathways), to incorporate the gene-gene as well as gene-drug regulatory relationships in synergic drug combination predictions. IDSP automatically learns weights of edges based on the gene and drug node relations, i.e., signaling interactions, by a multi-layer perceptron (MLP) and aggregates information in an inductive manner. The proposed architecture generates interpretable drug synergy prediction by detecting important signaling interactions, and can be implemented when the underlying molecular mechanism encounters unseen genes or signaling pathways. We test IDWSP on signaling networks formulated by genes from 46 core cancer signaling pathways and drug combinations from NCI ALMANAC drug combination screening data. The experimental results demonstrated that 1) IDSP can learn from the underlying molecular mechanism to make prediction without additional drug chemical information while achieving highly comparable performance with current state-of-art methods; 2) IDSP show superior generality and flexibility to implement the synergy prediction task on both transductive tasks and inductive tasks. 3) IDSP can generate interpretable results by detecting different salient signaling patterns (i.e. MoS) for different cell lines.