 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDART-Eval: A Comprehensive DNA Language Model Evaluation Benchmark on Regulatory DNA
Dec 06, 2024



Recent advances in self-supervised models for natural language, vision, and protein sequences have inspired the development of large genomic DNA language models (DNALMs). These models aim to learn generalizable representations of diverse DNA elements, potentially enabling various genomic prediction, interpretation and design tasks. Despite their potential, existing benchmarks do not adequately assess the capabilities of DNALMs on key downstream applications involving an important class of non-coding DNA elements critical for regulating gene activity. In this study, we introduce DART-Eval, a suite of representative benchmarks specifically focused on regulatory DNA to evaluate model performance across zero-shot, probed, and fine-tuned scenarios against contemporary ab initio models as baselines. Our benchmarks target biologically meaningful downstream tasks such as functional sequence feature discovery, predicting cell-type specific regulatory activity, and counterfactual prediction of the impacts of genetic variants. We find that current DNALMs exhibit inconsistent performance and do not offer compelling gains over alternative baseline models for most tasks, while requiring significantly more computational resources. We discuss potentially promising modeling, data curation, and evaluation strategies for the next generation of DNALMs. Our code is available at https://github.com/kundajelab/DART-Eval.
WILDS: A Benchmark of in-the-Wild Distribution Shifts
Dec 14, 2020
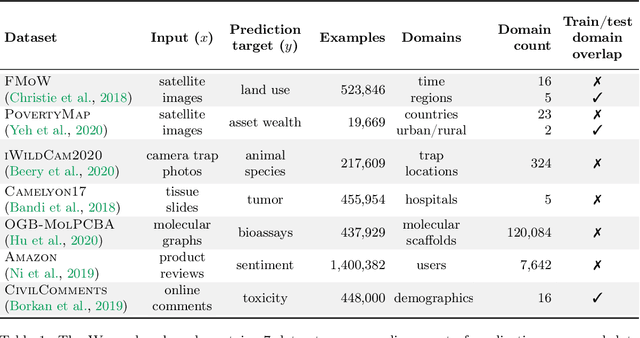

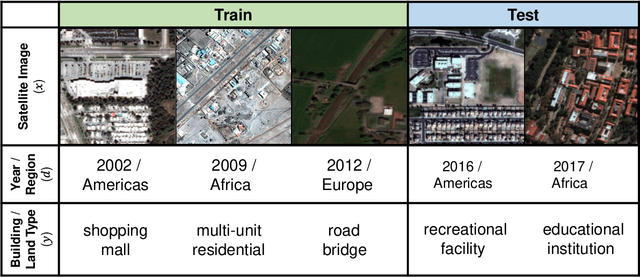
Distribution shifts can cause significant degradation in a broad range of machine learning (ML) systems deployed in the wild. However, many widely-used datasets in the ML community today were not designed for evaluating distribution shifts. These datasets typically have training and test sets drawn from the same distribution, and prior work on retrofitting them with distribution shifts has generally relied on artificial shifts that need not represent the kinds of shifts encountered in the wild. In this paper, we present WILDS, a benchmark of in-the-wild distribution shifts spanning diverse data modalities and applications, from tumor identification to wildlife monitoring to poverty mapping. WILDS builds on top of recent data collection efforts by domain experts in these applications and provides a unified collection of datasets with evaluation metrics and train/test splits that are representative of real-world distribution shifts. These datasets reflect distribution shifts arising from training and testing on different hospitals, cameras, countries, time periods, demographics, molecular scaffolds, etc., all of which cause substantial performance drops in our baseline models. Finally, we survey other applications that would be promising additions to the benchmark but for which we did not manage to find appropriate datasets; we discuss their associated challenges and detail datasets and shifts where we did not see an appreciable performance drop. By unifying datasets from a variety of application areas and making them accessible to the ML community, we hope to encourage the development of general-purpose methods that are anchored to real-world distribution shifts and that work well across different applications and problem settings. Data loaders, default models, and leaderboards are available at https://wilds.stanford.edu.
Calibration with Bias-Corrected Temperature Scaling Improves Domain Adaptation Under Label Shift in Modern Neural Networks
Jan 21, 2019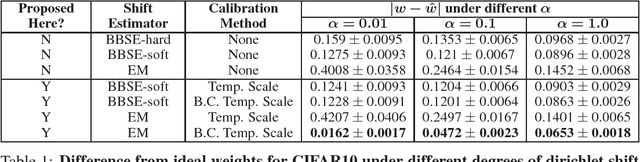
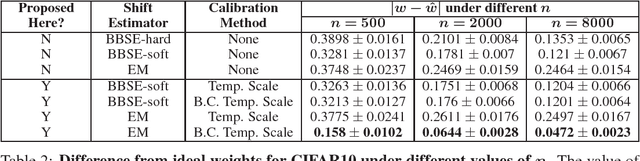
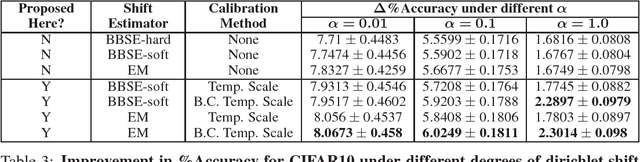
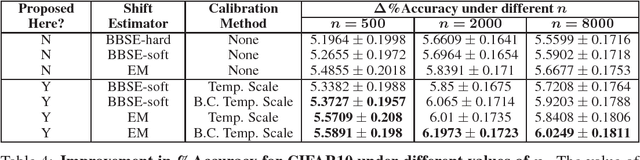
Label shift refers to the phenomenon where the marginal probability p(y) of observing a particular class changes between the training and test distributions while the conditional probability p(x|y) stays fixed. This is relevant in settings such as medical diagnosis, where a classifier trained to predict disease based on observed symptoms may need to be adapted to a different distribution where the baseline frequency of the disease is higher. Given calibrated estimates of p(y|x), one can apply an EM algorithm to correct for the shift in class imbalance between the training and test distributions without ever needing to calculate p(x|y). Unfortunately, modern neural networks typically fail to produce well-calibrated probabilities, compromising the effectiveness of this approach. Although Temperature Scaling can greatly reduce miscalibration in these networks, it can leave behind a systematic bias in the probabilities that still poses a problem. To address this, we extend Temperature Scaling with class-specific bias parameters, which largely eliminates systematic bias in the calibrated probabilities and allows for effective domain adaptation under label shift. We term our calibration approach "Bias-Corrected Temperature Scaling". On experiments with CIFAR10, we find that EM with Bias-Corrected Temperature Scaling significantly outperforms both EM with Temperature Scaling and the recently-proposed Black-Box Shift Estimation.
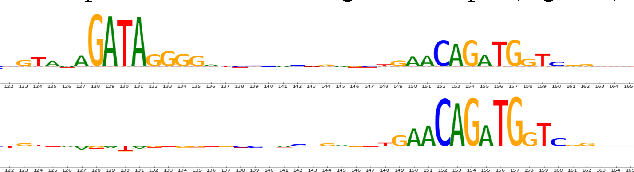
TF-MoDISco v0.4.4.2-alpha: Technical Note
Oct 31, 2018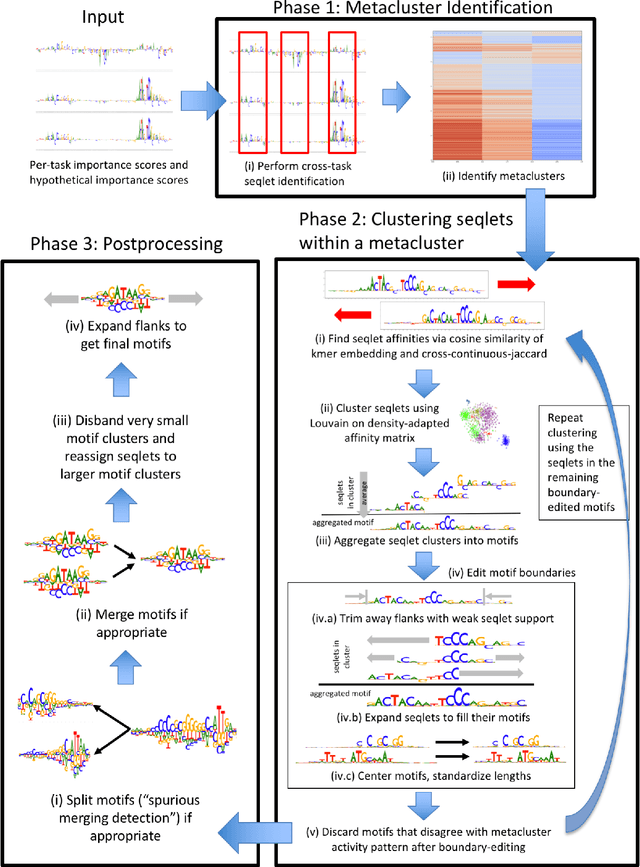
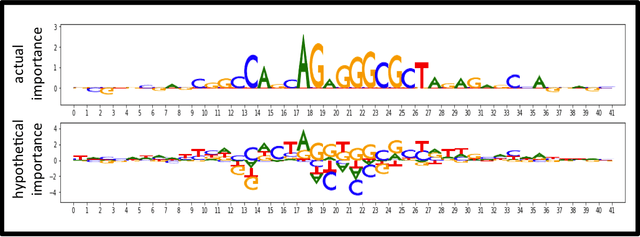
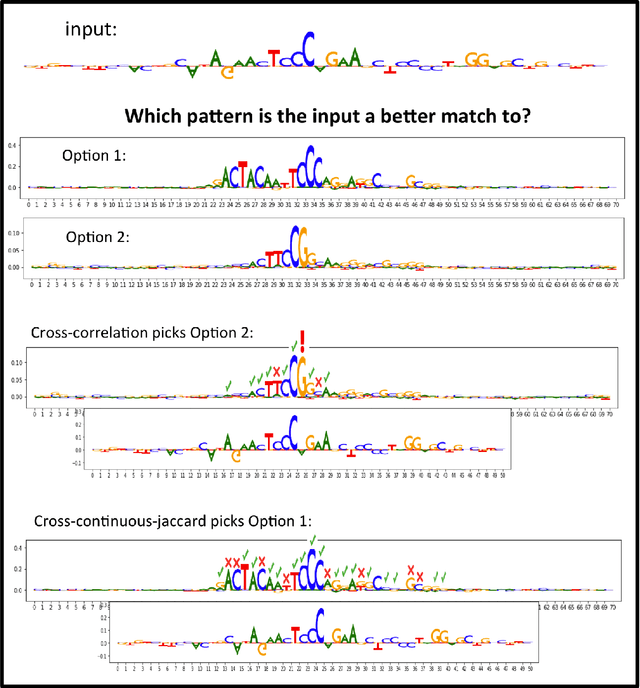
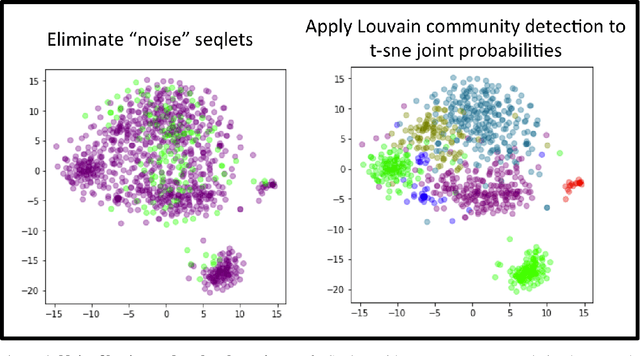
TF-MoDISco (Transcription Factor Motif Discovery from Importance Scores) is an algorithm for identifying motifs from basepair-level importance scores computed on genomic sequence data. This paper describes the methods behind TF-MoDISco version 0.4.4.2-alpha (available at https://github.com/kundajelab/tfmodisco/tree/v0.4.2.2-alpha).
Selective Classification via Curve Optimization
Sep 14, 2018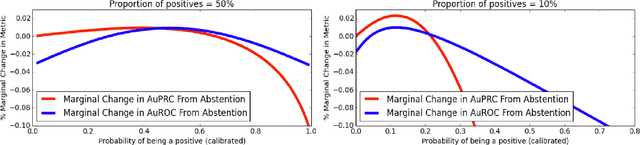
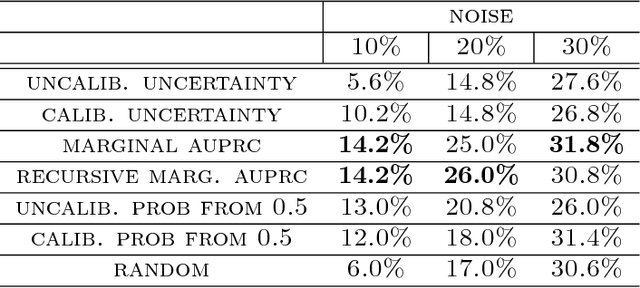
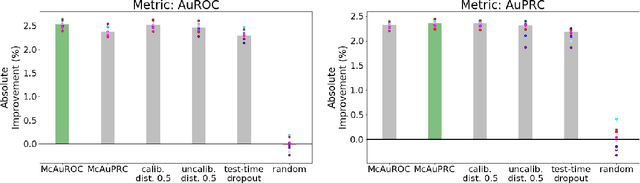
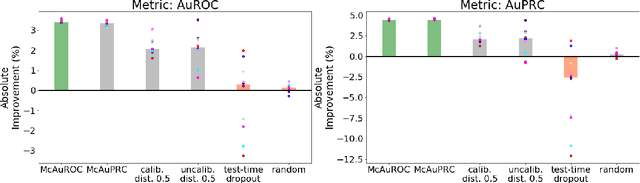
In practical applications of machine learning, it is often desirable to identify and abstain on examples where the model's predictions are likely to be incorrect. We consider the problem of selecting a budget-constrained subset of test examples to abstain on, with the goal of maximizing performance on the remaining examples. We develop a novel approach to this problem by analytically optimizing the expected marginal improvement in a desired performance metric, such as the area under the ROC curve or Precision-Recall curve. We compare our approach to other abstention techniques for deep learning models based on posterior probability and uncertainty estimates obtained using test-time dropout. On various tasks in computer vision, natural language processing, and bioinformatics, we demonstrate the consistent effectiveness of our approach over other techniques.
Computationally Efficient Measures of Internal Neuron Importance
Jul 26, 2018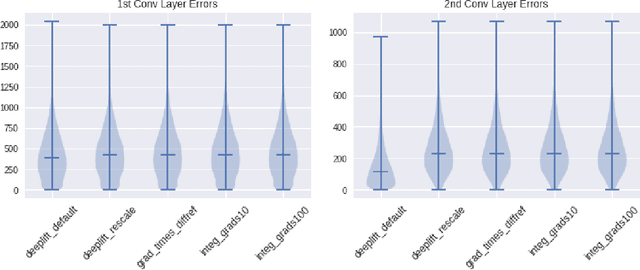
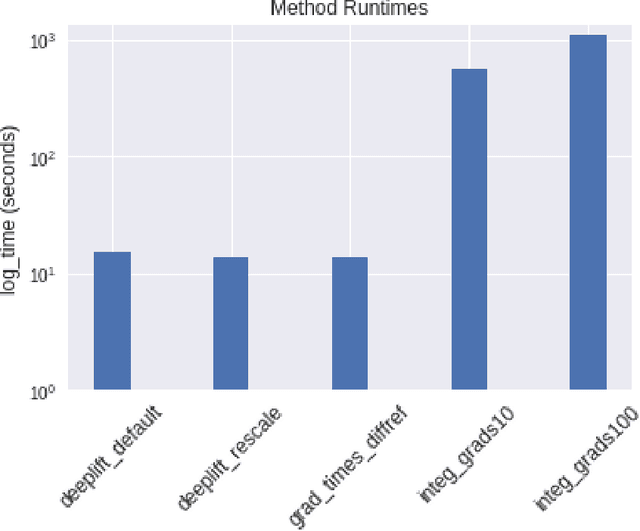
The challenge of assigning importance to individual neurons in a network is of interest when interpreting deep learning models. In recent work, Dhamdhere et al. proposed Total Conductance, a "natural refinement of Integrated Gradients" for attributing importance to internal neurons. Unfortunately, the authors found that calculating conductance in tensorflow required the addition of several custom gradient operators and did not scale well. In this work, we show that the formula for Total Conductance is mathematically equivalent to Path Integrated Gradients computed on a hidden layer in the network. We provide a scalable implementation of Total Conductance using standard tensorflow gradient operators that we call Neuron Integrated Gradients. We compare Neuron Integrated Gradients to DeepLIFT, a pre-existing computationally efficient approach that is applicable to calculating internal neuron importance. We find that DeepLIFT produces strong empirical results and is faster to compute, but because it lacks the theoretical properties of Neuron Integrated Gradients, it may not always be preferred in practice. Colab notebook reproducing results: http://bit.ly/neuronintegratedgradients
Not Just a Black Box: Learning Important Features Through Propagating Activation Differences
Apr 11, 2017
Note: This paper describes an older version of DeepLIFT. See https://arxiv.org/abs/1704.02685 for the newer version. Original abstract follows: The purported "black box" nature of neural networks is a barrier to adoption in applications where interpretability is essential. Here we present DeepLIFT (Learning Important FeaTures), an efficient and effective method for computing importance scores in a neural network. DeepLIFT compares the activation of each neuron to its 'reference activation' and assigns contribution scores according to the difference. We apply DeepLIFT to models trained on natural images and genomic data, and show significant advantages over gradient-based methods.
Learning Important Features Through Propagating Activation Differences
Apr 10, 2017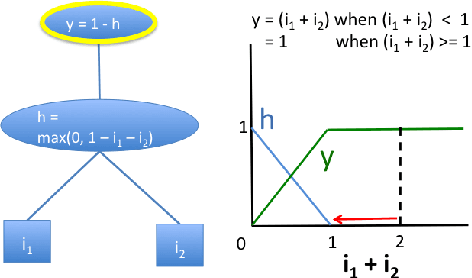
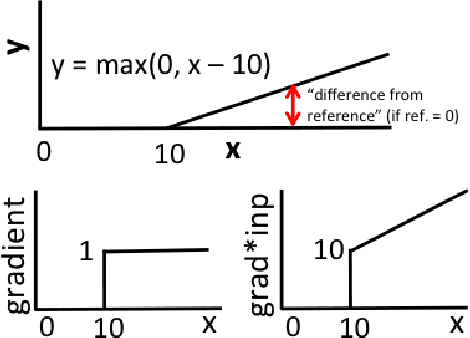
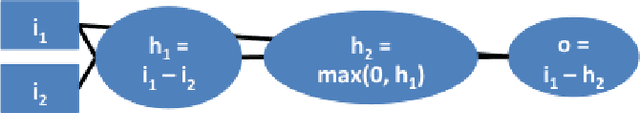
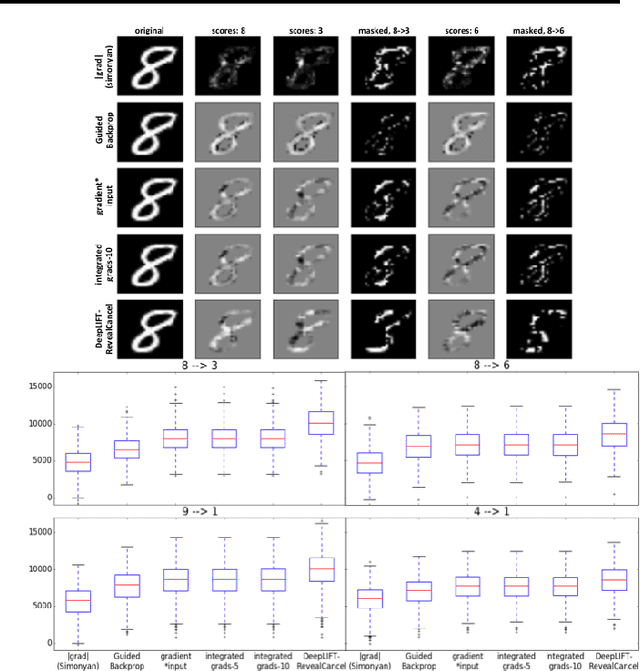
The purported "black box"' nature of neural networks is a barrier to adoption in applications where interpretability is essential. Here we present DeepLIFT (Deep Learning Important FeaTures), a method for decomposing the output prediction of a neural network on a specific input by backpropagating the contributions of all neurons in the network to every feature of the input. DeepLIFT compares the activation of each neuron to its 'reference activation' and assigns contribution scores according to the difference. By optionally giving separate consideration to positive and negative contributions, DeepLIFT can also reveal dependencies which are missed by other approaches. Scores can be computed efficiently in a single backward pass. We apply DeepLIFT to models trained on MNIST and simulated genomic data, and show significant advantages over gradient-based methods. A detailed video tutorial on the method is at http://goo.gl/qKb7pL and code is at http://goo.gl/RM8jvH.
* 9 pages, 6 figures