 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBayesian Optimization of Antibodies Informed by a Generative Model of Evolving Sequences
Dec 10, 2024To build effective therapeutics, biologists iteratively mutate antibody sequences to improve binding and stability. Proposed mutations can be informed by previous measurements or by learning from large antibody databases to predict only typical antibodies. Unfortunately, the space of typical antibodies is enormous to search, and experiments often fail to find suitable antibodies on a budget. We introduce Clone-informed Bayesian Optimization (CloneBO), a Bayesian optimization procedure that efficiently optimizes antibodies in the lab by teaching a generative model how our immune system optimizes antibodies. Our immune system makes antibodies by iteratively evolving specific portions of their sequences to bind their target strongly and stably, resulting in a set of related, evolving sequences known as a clonal family. We train a large language model, CloneLM, on hundreds of thousands of clonal families and use it to design sequences with mutations that are most likely to optimize an antibody within the human immune system. We propose to guide our designs to fit previous measurements with a twisted sequential Monte Carlo procedure. We show that CloneBO optimizes antibodies substantially more efficiently than previous methods in realistic in silico experiments and designs stronger and more stable binders in in vitro wet lab experiments.
Generative Humanization for Therapeutic Antibodies
Dec 09, 2024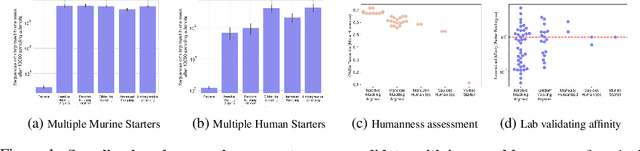
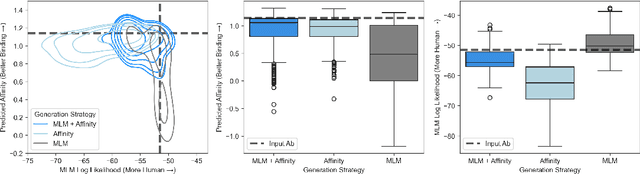
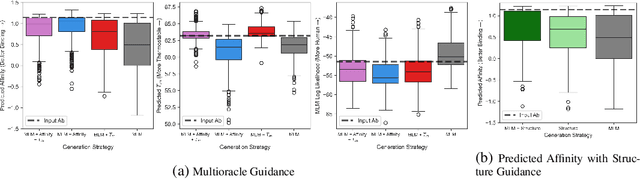
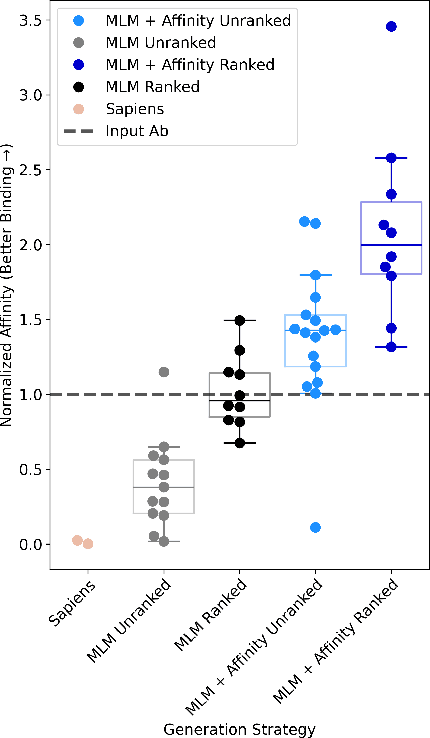
Antibody therapies have been employed to address some of today's most challenging diseases, but must meet many criteria during drug development before reaching a patient. Humanization is a sequence optimization strategy that addresses one critical risk called immunogenicity - a patient's immune response to the drug - by making an antibody more "human-like" in the absence of a predictive lab-based test for immunogenicity. However, existing humanization strategies generally yield very few humanized candidates, which may have degraded biophysical properties or decreased drug efficacy. Here, we re-frame humanization as a conditional generative modeling task, where humanizing mutations are sampled from a language model trained on human antibody data. We describe a sampling process that incorporates models of therapeutic attributes, such as antigen binding affinity, to obtain candidate sequences that have both reduced immunogenicity risk and maintained or improved therapeutic properties, allowing this algorithm to be readily embedded into an iterative antibody optimization campaign. We demonstrate in silico and in lab validation that in real therapeutic programs our generative humanization method produces diverse sets of antibodies that are both (1) highly-human and (2) have favorable therapeutic properties, such as improved binding to target antigens.
Accelerating Bayesian Optimization for Biological Sequence Design with Denoising Autoencoders
Mar 23, 2022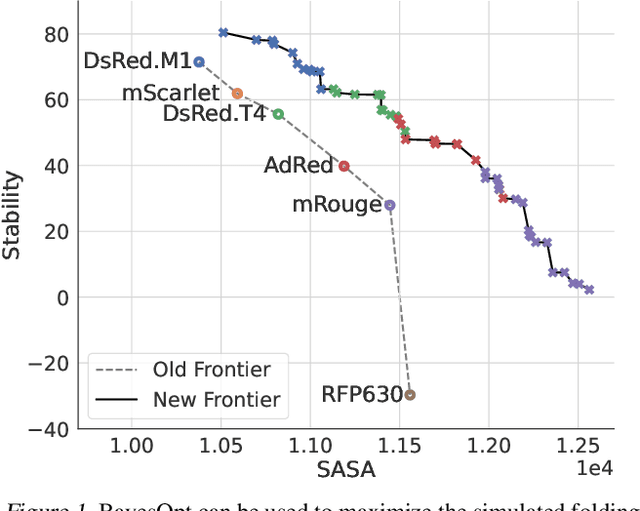
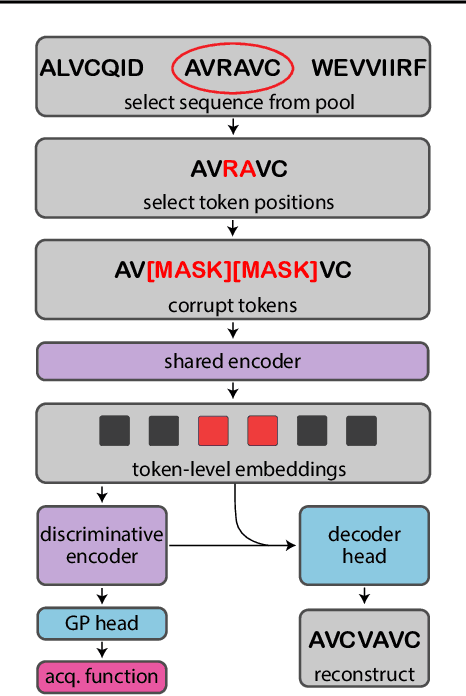
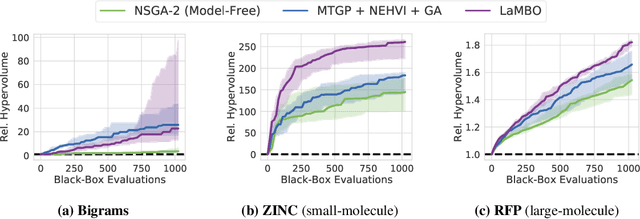
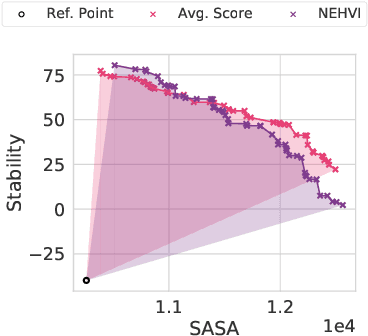
Bayesian optimization is a gold standard for query-efficient continuous optimization. However, its adoption for drug and antibody sequence design has been hindered by the discrete, high-dimensional nature of the decision variables. We develop a new approach (LaMBO) which jointly trains a denoising autoencoder with a discriminative multi-task Gaussian process head, enabling gradient-based optimization of multi-objective acquisition functions in the latent space of the autoencoder. These acquisition functions allow LaMBO to balance the explore-exploit trade-off over multiple design rounds, and to balance objective tradeoffs by optimizing sequences at many different points on the Pareto frontier. We evaluate LaMBO on a small-molecule task based on the ZINC dataset and introduce a new large-molecule task targeting fluorescent proteins. In our experiments, LaMBO outperforms genetic optimizers and does not require a large pretraining corpus, demonstrating that Bayesian optimization is practical and effective for biological sequence design.
A Hierarchical Approach to Scaling Batch Active Search Over Structured Data
Jul 20, 2020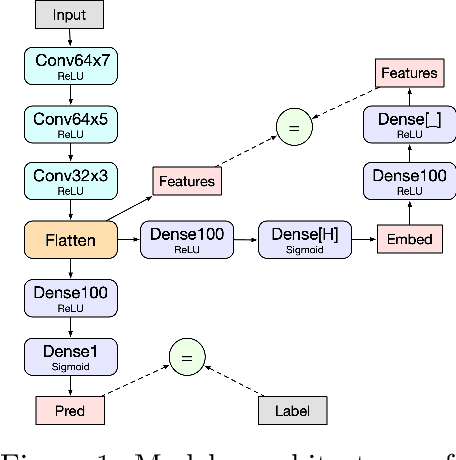

Active search is the process of identifying high-value data points in a large and often high-dimensional parameter space that can be expensive to evaluate. Traditional active search techniques like Bayesian optimization trade off exploration and exploitation over consecutive evaluations, and have historically focused on single or small (<5) numbers of examples evaluated per round. As modern data sets grow, so does the need to scale active search to large data sets and batch sizes. In this paper, we present a general hierarchical framework based on bandit algorithms to scale active search to large batch sizes by maximizing information derived from the unique structure of each dataset. Our hierarchical framework, Hierarchical Batch Bandit Search (HBBS), strategically distributes batch selection across a learned embedding space by facilitating wide exploration of different structural elements within a dataset. We focus our application of HBBS on modern biology, where large batch experimentation is often fundamental to the research process, and demonstrate batch design of biological sequences (protein and DNA). We also present a new Gym environment to easily simulate diverse biological sequences and to enable more comprehensive evaluation of active search methods across heterogeneous data sets. The HBBS framework improves upon standard performance, wall-clock, and scalability benchmarks for batch search by using a broad exploration strategy across coarse partitions and fine-grained exploitation within each partition of structured data.
Not Just a Black Box: Learning Important Features Through Propagating Activation Differences
Apr 11, 2017
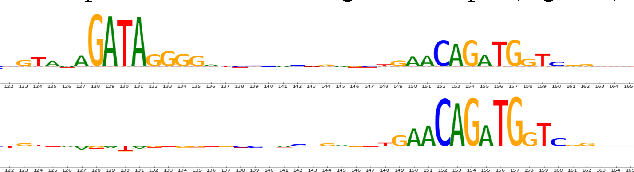
Note: This paper describes an older version of DeepLIFT. See https://arxiv.org/abs/1704.02685 for the newer version. Original abstract follows: The purported "black box" nature of neural networks is a barrier to adoption in applications where interpretability is essential. Here we present DeepLIFT (Learning Important FeaTures), an efficient and effective method for computing importance scores in a neural network. DeepLIFT compares the activation of each neuron to its 'reference activation' and assigns contribution scores according to the difference. We apply DeepLIFT to models trained on natural images and genomic data, and show significant advantages over gradient-based methods.
Learning Important Features Through Propagating Activation Differences
Apr 10, 2017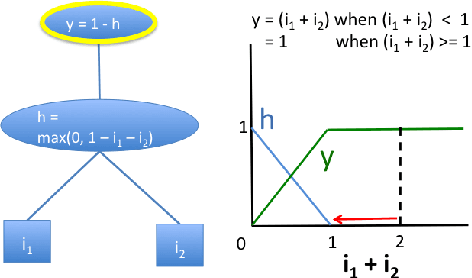
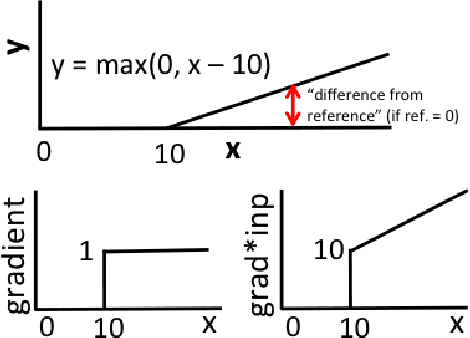
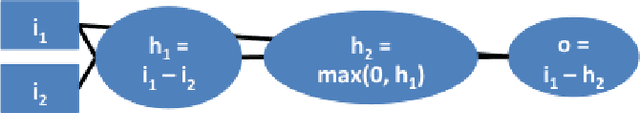
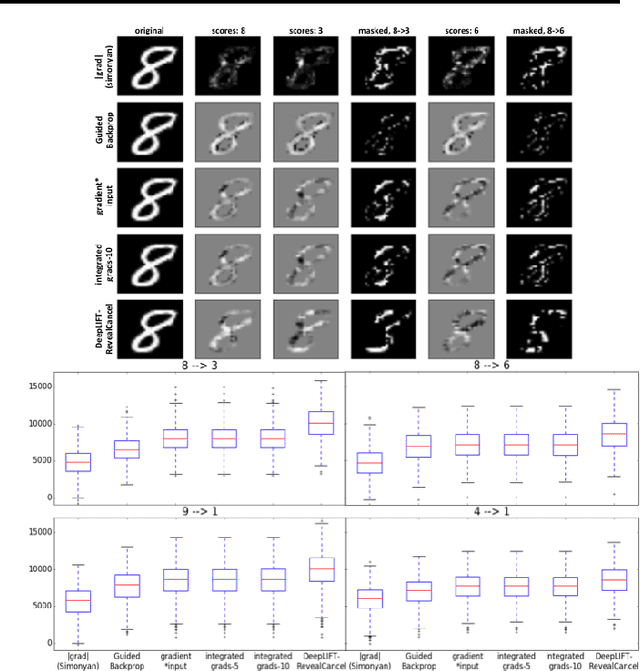
The purported "black box"' nature of neural networks is a barrier to adoption in applications where interpretability is essential. Here we present DeepLIFT (Deep Learning Important FeaTures), a method for decomposing the output prediction of a neural network on a specific input by backpropagating the contributions of all neurons in the network to every feature of the input. DeepLIFT compares the activation of each neuron to its 'reference activation' and assigns contribution scores according to the difference. By optionally giving separate consideration to positive and negative contributions, DeepLIFT can also reveal dependencies which are missed by other approaches. Scores can be computed efficiently in a single backward pass. We apply DeepLIFT to models trained on MNIST and simulated genomic data, and show significant advantages over gradient-based methods. A detailed video tutorial on the method is at http://goo.gl/qKb7pL and code is at http://goo.gl/RM8jvH.
* 9 pages, 6 figures