 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeContextBench: A Benchmark for Context Retrieval in Coding Agents
Feb 05, 2026LLM-based coding agents have shown strong performance on automated issue resolution benchmarks, yet existing evaluations largely focus on final task success, providing limited insight into how agents retrieve and use code context during problem solving. We introduce ContextBench, a process-oriented evaluation of context retrieval in coding agents. ContextBench consists of 1,136 issue-resolution tasks from 66 repositories across eight programming languages, each augmented with human-annotated gold contexts. We further implement an automated evaluation framework that tracks agent trajectories and measures context recall, precision, and efficiency throughout issue resolution. Using ContextBench, we evaluate four frontier LLMs and five coding agents. Our results show that sophisticated agent scaffolding yields only marginal gains in context retrieval ("The Bitter Lesson" of coding agents), LLMs consistently favor recall over precision, and substantial gaps exist between explored and utilized context. ContextBench augments existing end-to-end benchmarks with intermediate gold-context metrics that unbox the issue-resolution process. These contexts offer valuable intermediate signals for guiding LLM reasoning in software tasks. Data and code are available at: https://cioutn.github.io/context-bench/.
Bridging Code Graphs and Large Language Models for Better Code Understanding
Dec 08, 2025



Large Language Models (LLMs) have demonstrated remarkable performance in code intelligence tasks such as code generation, summarization, and translation. However, their reliance on linearized token sequences limits their ability to understand the structural semantics of programs. While prior studies have explored graphaugmented prompting and structure-aware pretraining, they either suffer from prompt length constraints or require task-specific architectural changes that are incompatible with large-scale instructionfollowing LLMs. To address these limitations, this paper proposes CGBridge, a novel plug-and-play method that enhances LLMs with Code Graph information through an external, trainable Bridge module. CGBridge first pre-trains a code graph encoder via selfsupervised learning on a large-scale dataset of 270K code graphs to learn structural code semantics. It then trains an external module to bridge the modality gap among code, graph, and text by aligning their semantics through cross-modal attention mechanisms. Finally, the bridge module generates structure-informed prompts, which are injected into a frozen LLM, and is fine-tuned for downstream code intelligence tasks. Experiments show that CGBridge achieves notable improvements over both the original model and the graphaugmented prompting method. Specifically, it yields a 16.19% and 9.12% relative gain in LLM-as-a-Judge on code summarization, and a 9.84% and 38.87% relative gain in Execution Accuracy on code translation. Moreover, CGBridge achieves over 4x faster inference than LoRA-tuned models, demonstrating both effectiveness and efficiency in structure-aware code understanding.
Wait, We Don't Need to "Wait"! Removing Thinking Tokens Improves Reasoning Efficiency
Jun 10, 2025Recent advances in large reasoning models have enabled complex, step-by-step reasoning but often introduce significant overthinking, resulting in verbose and redundant outputs that hinder efficiency. In this study, we examine whether explicit self-reflection, signaled by tokens such as "Wait" and "Hmm", is necessary for advanced reasoning. We propose NoWait, a simple yet effective approach that disables explicit self-reflection by suppressing these tokens during inference. Extensive experiments on ten benchmarks across textual, visual, and video reasoning tasks show that NoWait reduces chain-of-thought trajectory length by up to 27%-51% in five R1-style model series, without compromising model utility. NoWait thus offers a plug-and-play solution for efficient and utility-preserving multimodal reasoning.
CODESYNC: Synchronizing Large Language Models with Dynamic Code Evolution at Scale
Feb 23, 2025Large Language Models (LLMs) have exhibited exceptional performance in software engineering yet face challenges in adapting to continually evolving code knowledge, particularly regarding the frequent updates of third-party library APIs. This limitation, stemming from static pre-training datasets, often results in non-executable code or implementations with suboptimal safety and efficiency. To this end, this paper introduces CODESYNC, a data engine for identifying outdated code patterns and collecting real-time code knowledge updates from Python third-party libraries. Building upon CODESYNC, we develop CODESYNCBENCH, a comprehensive benchmark for assessing LLMs' ability to stay synchronized with code evolution, which covers real-world updates for 220 APIs from six Python libraries. Our benchmark offers 3,300 test cases across three evaluation tasks and an update-aware instruction tuning dataset consisting of 2,200 training samples. Extensive experiments on 14 state-of-the-art LLMs reveal that they struggle with dynamic code evolution, even with the support of advanced knowledge updating methods (e.g., DPO, ORPO, and SimPO). We believe that our benchmark can offer a strong foundation for the development of more effective methods for real-time code knowledge updating in the future. The experimental code and dataset are publicly available at: https://github.com/Lucky-voyage/Code-Sync.
How to Select Pre-Trained Code Models for Reuse? A Learning Perspective
Jan 07, 2025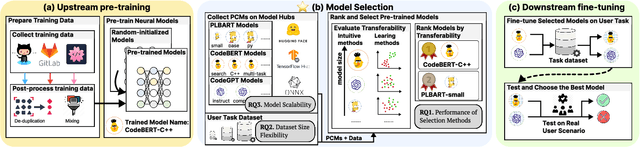
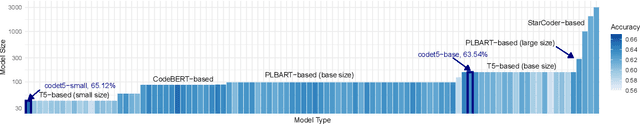
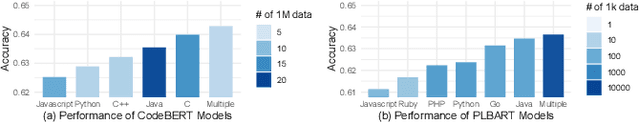
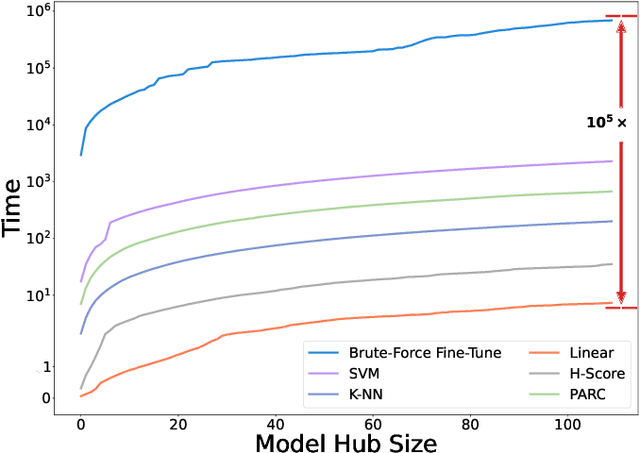
Pre-training a language model and then fine-tuning it has shown to be an efficient and effective technique for a wide range of code intelligence tasks, such as code generation, code summarization, and vulnerability detection. However, pretraining language models on a large-scale code corpus is computationally expensive. Fortunately, many off-the-shelf Pre-trained Code Models (PCMs), such as CodeBERT, CodeT5, CodeGen, and Code Llama, have been released publicly. These models acquire general code understanding and generation capability during pretraining, which enhances their performance on downstream code intelligence tasks. With an increasing number of these public pre-trained models, selecting the most suitable one to reuse for a specific task is essential. In this paper, we systematically investigate the reusability of PCMs. We first explore three intuitive model selection methods that select by size, training data, or brute-force fine-tuning. Experimental results show that these straightforward techniques either perform poorly or suffer high costs. Motivated by these findings, we explore learning-based model selection strategies that utilize pre-trained models without altering their parameters. Specifically, we train proxy models to gauge the performance of pre-trained models, and measure the distribution deviation between a model's latent features and the task's labels, using their closeness as an indicator of model transferability. We conduct experiments on 100 widely-used opensource PCMs for code intelligence tasks, with sizes ranging from 42.5 million to 3 billion parameters. The results demonstrate that learning-based selection methods reduce selection time to 100 seconds, compared to 2,700 hours with brute-force fine-tuning, with less than 6% performance degradation across related tasks.
Graph Neural Networks for Vulnerability Detection: A Counterfactual Explanation
Apr 24, 2024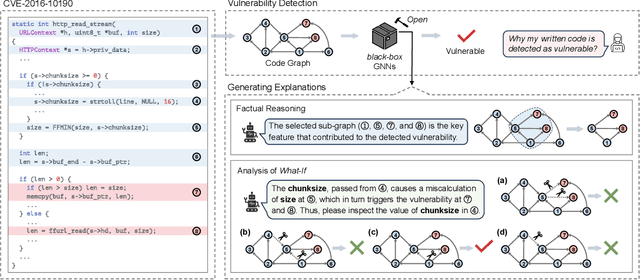
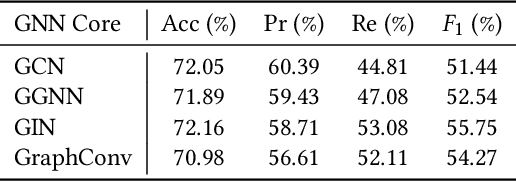
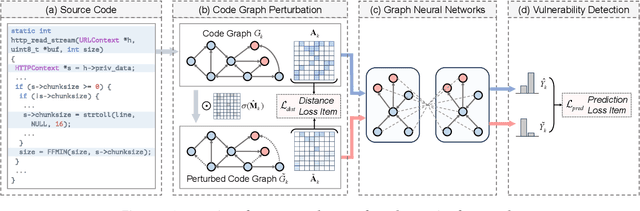
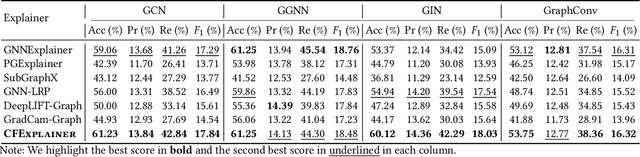
Vulnerability detection is crucial for ensuring the security and reliability of software systems. Recently, Graph Neural Networks (GNNs) have emerged as a prominent code embedding approach for vulnerability detection, owing to their ability to capture the underlying semantic structure of source code. However, GNNs face significant challenges in explainability due to their inherently black-box nature. To this end, several factual reasoning-based explainers have been proposed. These explainers provide explanations for the predictions made by GNNs by analyzing the key features that contribute to the outcomes. We argue that these factual reasoning-based explanations cannot answer critical what-if questions: What would happen to the GNN's decision if we were to alter the code graph into alternative structures? Inspired by advancements of counterfactual reasoning in artificial intelligence, we propose CFExplainer, a novel counterfactual explainer for GNN-based vulnerability detection. Unlike factual reasoning-based explainers, CFExplainer seeks the minimal perturbation to the input code graph that leads to a change in the prediction, thereby addressing the what-if questions for vulnerability detection. We term this perturbation a counterfactual explanation, which can pinpoint the root causes of the detected vulnerability and furnish valuable insights for developers to undertake appropriate actions for fixing the vulnerability. Extensive experiments on four GNN-based vulnerability detection models demonstrate the effectiveness of CFExplainer over existing state-of-the-art factual reasoning-based explainers.
Hierarchical Graph Representation Learning for the Prediction of Drug-Target Binding Affinity
Mar 22, 2022



The identification of drug-target binding affinity (DTA) has attracted increasing attention in the drug discovery process due to the more specific interpretation than binary interaction prediction. Recently, numerous deep learning-based computational methods have been proposed to predict the binding affinities between drugs and targets benefiting from their satisfactory performance. However, the previous works mainly focus on encoding biological features and chemical structures of drugs and targets, with a lack of exploiting the essential topological information from the drug-target affinity network. In this paper, we propose a novel hierarchical graph representation learning model for the drug-target binding affinity prediction, namely HGRL-DTA. The main contribution of our model is to establish a hierarchical graph learning architecture to incorporate the intrinsic properties of drug/target molecules and the topological affinities of drug-target pairs. In this architecture, we adopt a message broadcasting mechanism to integrate the hierarchical representations learned from the global-level affinity graph and the local-level molecular graph. Besides, we design a similarity-based embedding map to solve the cold start problem of inferring representations for unseen drugs and targets. Comprehensive experimental results under different scenarios indicate that HGRL-DTA significantly outperforms the state-of-the-art models and shows better model generalization among all the scenarios.