 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFast Bayesian Optimization of Needle-in-a-Haystack Problems using Zooming Memory-Based Initialization
Aug 26, 2022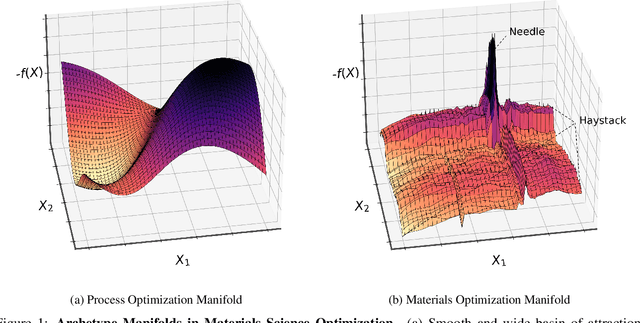
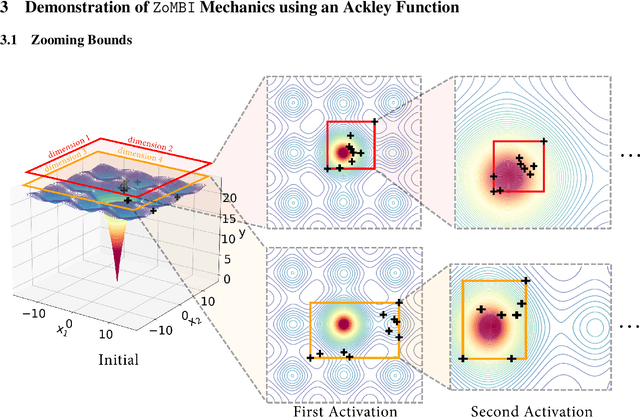
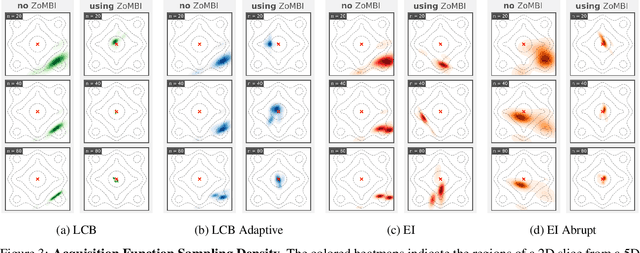
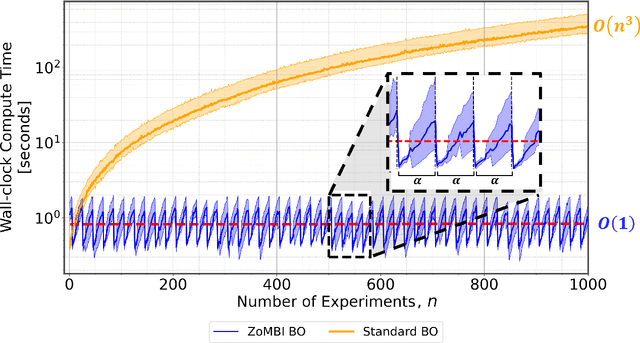
Needle-in-a-Haystack problems exist across a wide range of applications including rare disease prediction, ecological resource management, fraud detection, and material property optimization. A Needle-in-a-Haystack problem arises when there is an extreme imbalance of optimum conditions relative to the size of the dataset. For example, only 0.82% out of 146k total materials in the open-access Materials Project database have a negative Poisson's ratio. However, current state-of-the-art optimization algorithms are not designed with the capabilities to find solutions to these challenging multidimensional Needle-in-a-Haystack problems, resulting in slow convergence to a global optimum or pigeonholing into a local minimum. In this paper, we present a Zooming Memory-Based Initialization algorithm, entitled ZoMBI, that builds on conventional Bayesian optimization principles to quickly and efficiently optimize Needle-in-a-Haystack problems in both less time and fewer experiments by addressing the common convergence and pigeonholing issues. ZoMBI actively extracts knowledge from the previously best-performing evaluated experiments to iteratively zoom in the sampling search bounds towards the global optimum "needle" and then prunes the memory of low-performing historical experiments to accelerate compute times. We validate the algorithm's performance on two real-world 5-dimensional Needle-in-a-Haystack material property optimization datasets: discovery of auxetic Poisson's ratio materials and discovery of high thermoelectric figure of merit materials. The ZoMBI algorithm demonstrates compute time speed-ups of 400x compared to traditional Bayesian optimization as well as efficiently discovering materials in under 100 experiments that are up to 3x more highly optimized than those discovered by current state-of-the-art algorithms.
Machine Learning with Knowledge Constraints for Process Optimization of Open-Air Perovskite Solar Cell Manufacturing
Oct 15, 2021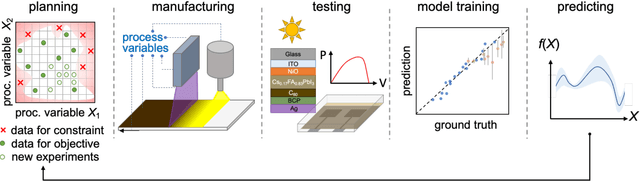
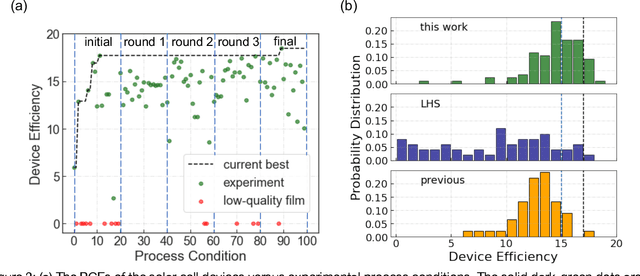
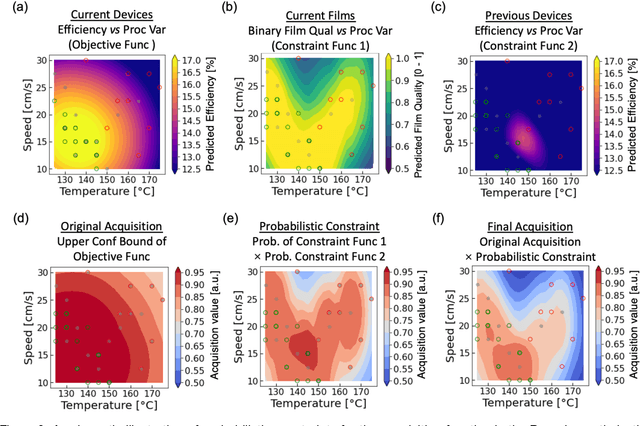
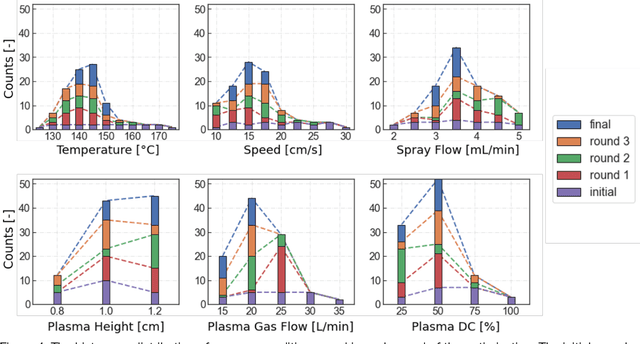
Perovskite photovoltaics (PV) have achieved rapid development in the past decade in terms of power conversion efficiency of small-area lab-scale devices; however, successful commercialization still requires further development of low-cost, scalable, and high-throughput manufacturing techniques. One of the key challenges to the development of a new fabrication technique is the high-dimensional parameter space, and machine learning (ML) can be used to accelerate perovskite PV scaling. Here, we present an ML-guided framework of sequential learning for manufacturing process optimization. We apply our methodology to the Rapid Spray Plasma Processing (RSPP) technique for perovskite thin films in ambient conditions. With a limited experimental budget of screening 100 conditions process conditions, we demonstrated an efficiency improvement to 18.5% for the best device, and we also experimentally found 10 unique conditions to produce the top-performing devices of more than 17% efficiency, which is 5 times higher rate of success than pseudo-random Latin hypercube sampling. Our model is enabled by three innovations: (a) flexible knowledge transfer between experimental processes by incorporating data from prior experimental data as a soft constraint; (b) incorporation of both subjective human observations and ML insights when selecting next experiments; (c) adaptive strategy of locating the region of interest using Bayesian optimization first, and then conducting local exploration for high-efficiency devices. Furthermore, in virtual benchmarking, our framework achieves faster improvements with limited experimental budgets than traditional design-of-experiments methods (e.g., one-variable-at-a-time sampling).
Benchmarking the Performance of Bayesian Optimization across Multiple Experimental Materials Science Domains
May 23, 2021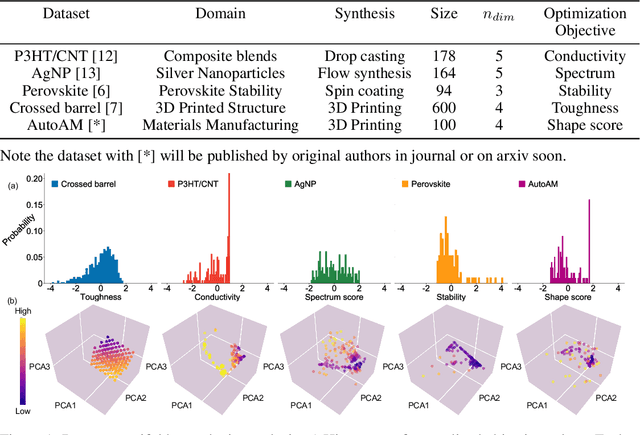
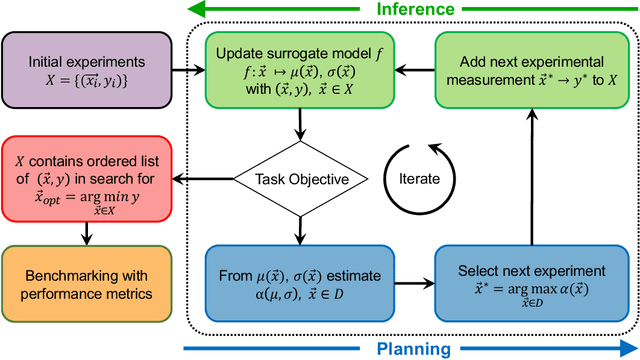
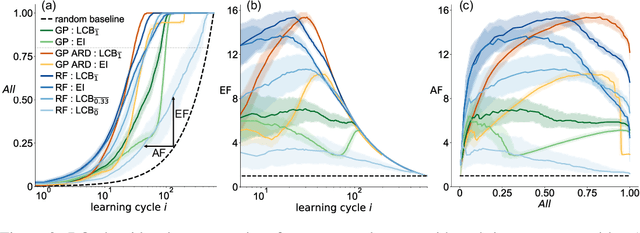
In the field of machine learning (ML) for materials optimization, active learning algorithms, such as Bayesian Optimization (BO), have been leveraged for guiding autonomous and high-throughput experimentation systems. However, very few studies have evaluated the efficiency of BO as a general optimization algorithm across a broad range of experimental materials science domains. In this work, we evaluate the performance of BO algorithms with a collection of surrogate model and acquisition function pairs across five diverse experimental materials systems, namely carbon nanotube polymer blends, silver nanoparticles, lead-halide perovskites, as well as additively manufactured polymer structures and shapes. By defining acceleration and enhancement metrics for general materials optimization objectives, we find that for surrogate model selection, Gaussian Process (GP) with anisotropic kernels (automatic relevance detection, ARD) and Random Forests (RF) have comparable performance and both outperform the commonly used GP without ARD. We discuss the implicit distributional assumptions of RF and GP, and the benefits of using GP with anisotropic kernels in detail. We provide practical insights for experimentalists on surrogate model selection of BO during materials optimization campaigns.
Inverse design of crystals using generalized invertible crystallographic representation
May 15, 2020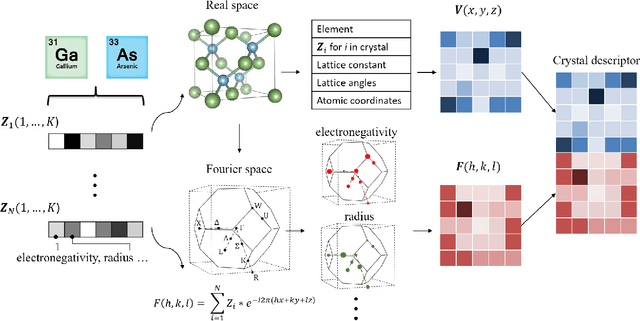
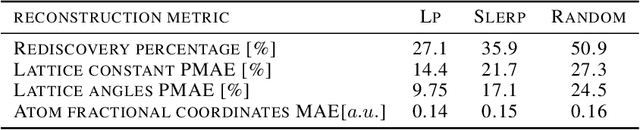
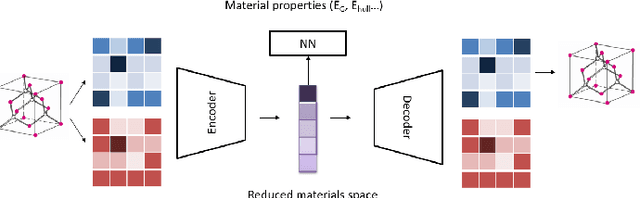
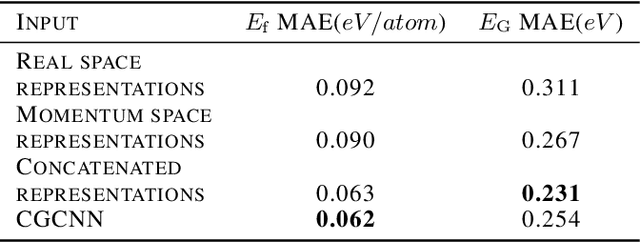
Deep learning has fostered many novel applications in materials informatics. However, the inverse design of inorganic crystals, $\textit{i.e.}$ generating new crystal structure with targeted properties, remains a grand challenge. An important ingredient for such generative models is an invertible representation that accesses the full periodic table. This is challenging due to limited data availability and the complexity of 3D periodic crystal structures. In this paper, we present a generalized invertible representation that encodes the crystallographic information into the descriptors in both real space and reciprocal space. Combining with a generative variational autoencoder (VAE), a wide range of crystallographic structures and chemistries with desired properties can be inverse-designed. We show that our VAE model predicts novel crystal structures that do not exist in the training and test database (Materials Project) with targeted formation energies and band gaps. We validate those predicted crystals by first-principles calculations. Finally, to design solids with practical applications, we address the sparse label problem by building a semi-supervised VAE and demonstrate its successful prediction of unique thermoelectric materials
Fast classification of small X-ray diffraction datasets using data augmentation and deep neural networks
Nov 20, 2018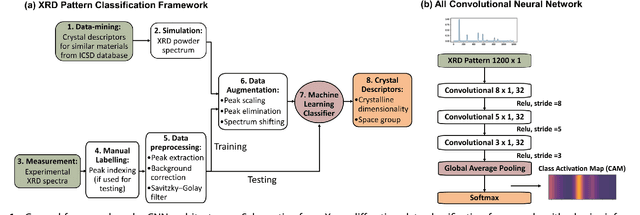
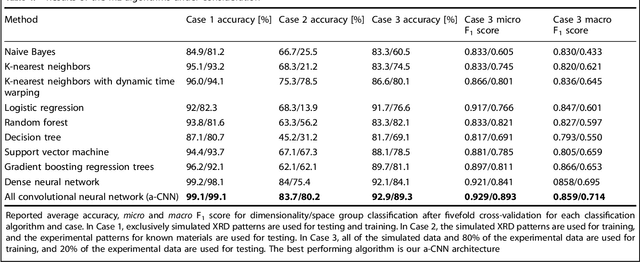
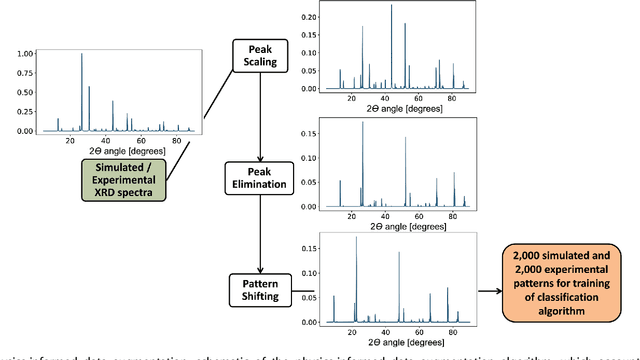
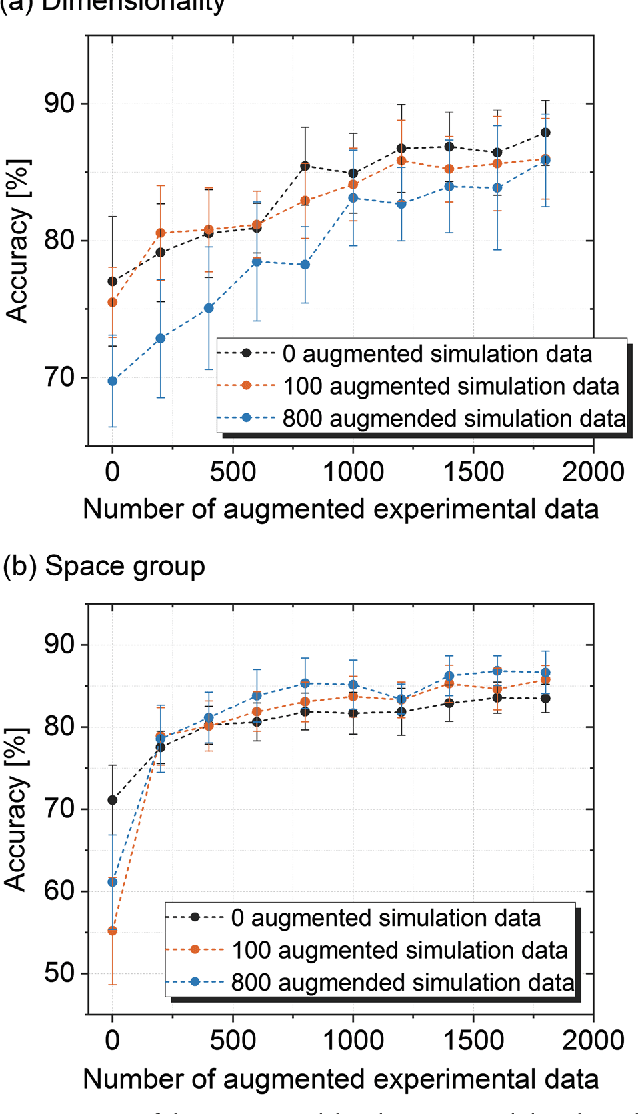
X-ray diffraction (XRD) for crystal structure characterization is among the most time-consuming and complex steps in the development cycle of novel materials. We propose a machine-learning-enabled approach to predict crystallographic dimensionality and space group from a limited number of experimental thin-film XRD patterns. We overcome the sparse-data problem intrinsic to novel materials development by coupling a supervised machine-learning approach with a physics-based data augmentation strategy . Using this approach, XRD spectrum acquisition and analysis occurs under 5.5 minutes, with accuracy comparable to human expert labeling. We simulate experimental powder diffraction patterns from crystallographic information contained in the Inorganic Crystal Structure Database (ICSD). We train a classification algorithm using a combination of labeled simulated and experimental augmented datasets, which account for thin-film characteristics and measurement noise. As a test case, 88 metal-halide thin films spanning 3 dimensionalities and 7 space-groups are synthesized and classified. The accuracies and throughputs of multiple machine-learning techniques are evaluated, along with the effect of augmented dataset size. The most accurate classification algorithm is found to be a feed-forward deep neural network. The calculated accuracies for dimensionality and space-group classification are comparable to ground-truth labelling by a human expert, approximately 90\% and 85\%, respectively. Additionally, we systematically evaluate the maximum XRD spectrum step size (data acquisition rate) before loss of predictive accuracy occurs, and determine it to be \ang{0.16} $2\theta $, which enables an XRD spectrum to be obtained and analyzed in 5 minutes or less.