 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFast classification of small X-ray diffraction datasets using data augmentation and deep neural networks
Paper and Code
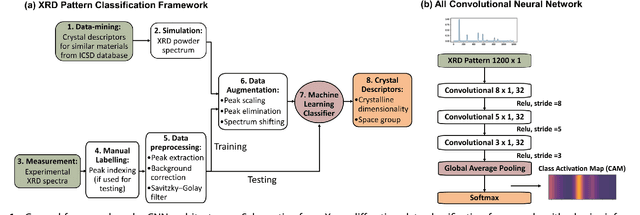
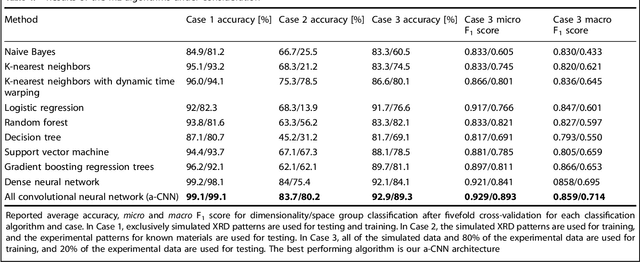
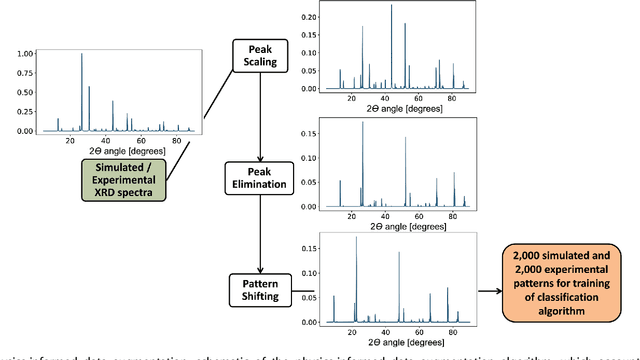
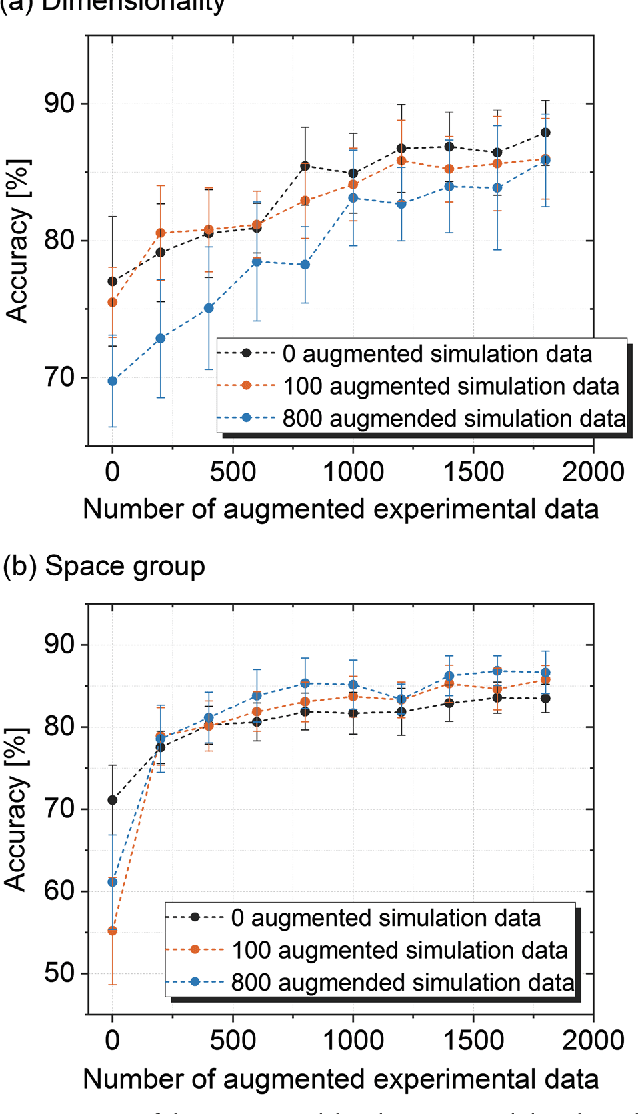
X-ray diffraction (XRD) for crystal structure characterization is among the most time-consuming and complex steps in the development cycle of novel materials. We propose a machine-learning-enabled approach to predict crystallographic dimensionality and space group from a limited number of experimental thin-film XRD patterns. We overcome the sparse-data problem intrinsic to novel materials development by coupling a supervised machine-learning approach with a physics-based data augmentation strategy . Using this approach, XRD spectrum acquisition and analysis occurs under 5.5 minutes, with accuracy comparable to human expert labeling. We simulate experimental powder diffraction patterns from crystallographic information contained in the Inorganic Crystal Structure Database (ICSD). We train a classification algorithm using a combination of labeled simulated and experimental augmented datasets, which account for thin-film characteristics and measurement noise. As a test case, 88 metal-halide thin films spanning 3 dimensionalities and 7 space-groups are synthesized and classified. The accuracies and throughputs of multiple machine-learning techniques are evaluated, along with the effect of augmented dataset size. The most accurate classification algorithm is found to be a feed-forward deep neural network. The calculated accuracies for dimensionality and space-group classification are comparable to ground-truth labelling by a human expert, approximately 90\% and 85\%, respectively. Additionally, we systematically evaluate the maximum XRD spectrum step size (data acquisition rate) before loss of predictive accuracy occurs, and determine it to be \ang{0.16} $2\theta $, which enables an XRD spectrum to be obtained and analyzed in 5 minutes or less.