 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRCDN: Real-Centered Detection Network for Robust Face Forgery Identification
Jan 17, 2026Image forgery has become a critical threat with the rapid proliferation of AI-based generation tools, which make it increasingly easy to synthesize realistic but fraudulent facial content. Existing detection methods achieve near-perfect performance when training and testing are conducted within the same domain, yet their effectiveness deteriorates substantially in crossdomain scenarios. This limitation is problematic, as new forgery techniques continuously emerge and detectors must remain reliable against unseen manipulations. To address this challenge, we propose the Real-Centered Detection Network (RCDN), a frequency spatial convolutional neural networks(CNN) framework with an Xception backbone that anchors its representation space around authentic facial images. Instead of modeling the diverse and evolving patterns of forgeries, RCDN emphasizes the consistency of real images, leveraging a dual-branch architecture and a real centered loss design to enhance robustness under distribution shifts. Extensive experiments on the DiFF dataset, focusing on three representative forgery types (FE, I2I, T2I), demonstrate that RCDN achieves both state-of-the-art in-domain accuracy and significantly stronger cross-domain generalization. Notably, RCDN reduces the generalization gap compared to leading baselines and achieves the highest cross/in-domain stability ratio, highlighting its potential as a practical solution for defending against evolving and unseen image forgery techniques.
Deep Learning for Taxol Exposure Analysis: A New Cell Image Dataset and Attention-Based Baseline Model
Aug 20, 2025Monitoring the effects of the chemotherapeutic agent Taxol at the cellular level is critical for both clinical evaluation and biomedical research. However, existing detection methods require specialized equipment, skilled personnel, and extensive sample preparation, making them expensive, labor-intensive, and unsuitable for high-throughput or real-time analysis. Deep learning approaches have shown great promise in medical and biological image analysis, enabling automated, high-throughput assessment of cellular morphology. Yet, no publicly available dataset currently exists for automated morphological analysis of cellular responses to Taxol exposure. To address this gap, we introduce a new microscopy image dataset capturing C6 glioma cells treated with varying concentrations of Taxol. To provide an effective solution for Taxol concentration classification and establish a benchmark for future studies on this dataset, we propose a baseline model named ResAttention-KNN, which combines a ResNet-50 with Convolutional Block Attention Modules and uses a k-Nearest Neighbors classifier in the learned embedding space. This model integrates attention-based refinement and non-parametric classification to enhance robustness and interpretability. Both the dataset and implementation are publicly released to support reproducibility and facilitate future research in vision-based biomedical analysis.
Revising the Problem of Partial Labels from the Perspective of CNNs' Robustness
Jul 24, 2024
Convolutional neural networks (CNNs) have gained increasing popularity and versatility in recent decades, finding applications in diverse domains. These remarkable achievements are greatly attributed to the support of extensive datasets with precise labels. However, annotating image datasets is intricate and complex, particularly in the case of multi-label datasets. Hence, the concept of partial-label setting has been proposed to reduce annotation costs, and numerous corresponding solutions have been introduced. The evaluation methods for these existing solutions have been primarily based on accuracy. That is, their performance is assessed by their predictive accuracy on the test set. However, we insist that such an evaluation is insufficient and one-sided. On one hand, since the quality of the test set has not been evaluated, the assessment results are unreliable. On the other hand, the partial-label problem may also be raised by undergoing adversarial attacks. Therefore, incorporating robustness into the evaluation system is crucial. For this purpose, we first propose two attack models to generate multiple partial-label datasets with varying degrees of label missing rates. Subsequently, we introduce a lightweight partial-label solution using pseudo-labeling techniques and a designed loss function. Then, we employ D-Score to analyze both the proposed and existing methods to determine whether they can enhance robustness while improving accuracy. Extensive experimental results demonstrate that while certain methods may improve accuracy, the enhancement in robustness is not significant, and in some cases, it even diminishes.
AlphaCrystal-II: Distance matrix based crystal structure prediction using deep learning
Apr 07, 2024



Computational prediction of stable crystal structures has a profound impact on the large-scale discovery of novel functional materials. However, predicting the crystal structure solely from a material's composition or formula is a promising yet challenging task, as traditional ab initio crystal structure prediction (CSP) methods rely on time-consuming global searches and first-principles free energy calculations. Inspired by the recent success of deep learning approaches in protein structure prediction, which utilize pairwise amino acid interactions to describe 3D structures, we present AlphaCrystal-II, a novel knowledge-based solution that exploits the abundant inter-atomic interaction patterns found in existing known crystal structures. AlphaCrystal-II predicts the atomic distance matrix of a target crystal material and employs this matrix to reconstruct its 3D crystal structure. By leveraging the wealth of inter-atomic relationships of known crystal structures, our approach demonstrates remarkable effectiveness and reliability in structure prediction through comprehensive experiments. This work highlights the potential of data-driven methods in accelerating the discovery and design of new materials with tailored properties.
Towards Imbalanced Large Scale Multi-label Classification with Partially Annotated Labels
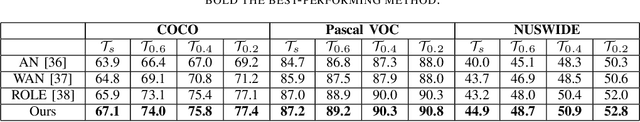
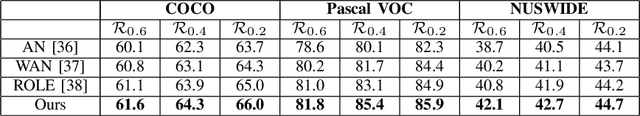
Jul 31, 2023Multi-label classification is a widely encountered problem in daily life, where an instance can be associated with multiple classes. In theory, this is a supervised learning method that requires a large amount of labeling. However, annotating data is time-consuming and may be infeasible for huge labeling spaces. In addition, label imbalance can limit the performance of multi-label classifiers, especially when some labels are missing. Therefore, it is meaningful to study how to train neural networks using partial labels. In this work, we address the issue of label imbalance and investigate how to train classifiers using partial labels in large labeling spaces. First, we introduce the pseudo-labeling technique, which allows commonly adopted networks to be applied in partially labeled settings without the need for additional complex structures. Then, we propose a novel loss function that leverages statistical information from existing datasets to effectively alleviate the label imbalance problem. In addition, we design a dynamic training scheme to reduce the dimension of the labeling space and further mitigate the imbalance. Finally, we conduct extensive experiments on some publicly available multi-label datasets such as COCO, NUS-WIDE, CUB, and Open Images to demonstrate the effectiveness of the proposed approach. The results show that our approach outperforms several state-of-the-art methods, and surprisingly, in some partial labeling settings, our approach even exceeds the methods trained with full labels.
D-Score: A White-Box Diagnosis Score for CNNs Based on Mutation Operators
Apr 03, 2023Convolutional neural networks (CNNs) have been widely applied in many safety-critical domains, such as autonomous driving and medical diagnosis. However, concerns have been raised with respect to the trustworthiness of these models: The standard testing method evaluates the performance of a model on a test set, while low-quality and insufficient test sets can lead to unreliable evaluation results, which can have unforeseeable consequences. Therefore, how to comprehensively evaluate CNNs and, based on the evaluation results, how to enhance their trustworthiness are the key problems to be urgently addressed. Prior work has used mutation tests to evaluate the test sets of CNNs. However, the evaluation scores are black boxes and not explicit enough for what is being tested. In this paper, we propose a white-box diagnostic approach that uses mutation operators and image transformation to calculate the feature and attention distribution of the model and further present a diagnosis score, namely D-Score, to reflect the model's robustness and fitness to a dataset. We also propose a D-Score based data augmentation method to enhance the CNN's performance to translations and rescalings. Comprehensive experiments on two widely used datasets and three commonly adopted CNNs demonstrate the effectiveness of our approach.
Discovery of 2D materials using Transformer Network based Generative Design
Jan 14, 2023Two-dimensional (2D) materials have wide applications in superconductors, quantum, and topological materials. However, their rational design is not well established, and currently less than 6,000 experimentally synthesized 2D materials have been reported. Recently, deep learning, data-mining, and density functional theory (DFT)-based high-throughput calculations are widely performed to discover potential new materials for diverse applications. Here we propose a generative material design pipeline, namely material transformer generator(MTG), for large-scale discovery of hypothetical 2D materials. We train two 2D materials composition generators using self-learning neural language models based on Transformers with and without transfer learning. The models are then used to generate a large number of candidate 2D compositions, which are fed to known 2D materials templates for crystal structure prediction. Next, we performed DFT computations to study their thermodynamic stability based on energy-above-hull and formation energy. We report four new DFT-verified stable 2D materials with zero e-above-hull energies, including NiCl$_4$, IrSBr, CuBr$_3$, and CoBrCl. Our work thus demonstrates the potential of our MTG generative materials design pipeline in the discovery of novel 2D materials and other functional materials.
Depth Monocular Estimation with Attention-based Encoder-Decoder Network from Single Image
Oct 24, 2022



Depth information is the foundation of perception, essential for autonomous driving, robotics, and other source-constrained applications. Promptly obtaining accurate and efficient depth information allows for a rapid response in dynamic environments. Sensor-based methods using LIDAR and RADAR obtain high precision at the cost of high power consumption, price, and volume. While due to advances in deep learning, vision-based approaches have recently received much attention and can overcome these drawbacks. In this work, we explore an extreme scenario in vision-based settings: estimate a depth map from one monocular image severely plagued by grid artifacts and blurry edges. To address this scenario, We first design a convolutional attention mechanism block (CAMB) which consists of channel attention and spatial attention sequentially and insert these CAMBs into skip connections. As a result, our novel approach can find the focus of current image with minimal overhead and avoid losses of depth features. Next, by combining the depth value, the gradients of X axis, Y axis and diagonal directions, and the structural similarity index measure (SSIM), we propose our novel loss function. Moreover, we utilize pixel blocks to accelerate the computation of the loss function. Finally, we show, through comprehensive experiments on two large-scale image datasets, i.e. KITTI and NYU-V2, that our method outperforms several representative baselines.
An Effective Approach for Multi-label Classification with Missing Labels
Oct 24, 2022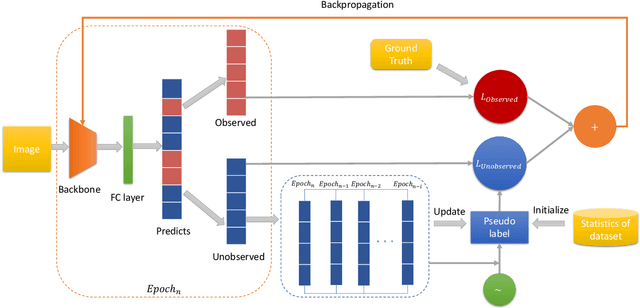
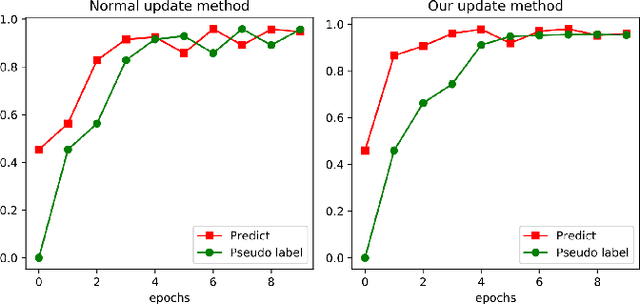
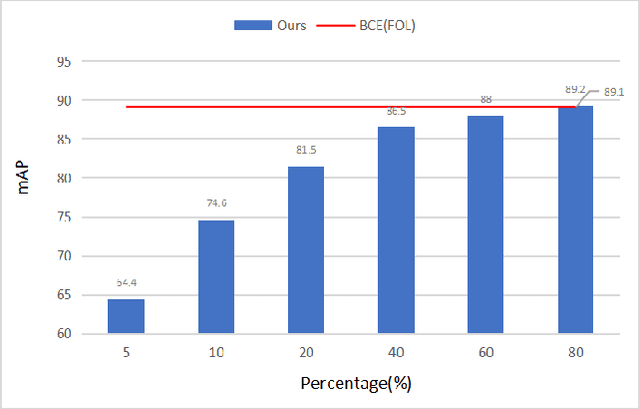
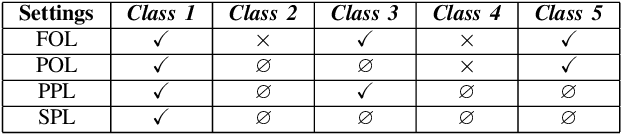
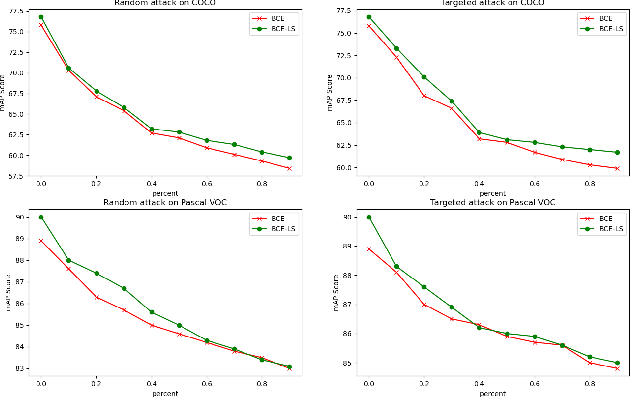
Compared with multi-class classification, multi-label classification that contains more than one class is more suitable in real life scenarios. Obtaining fully labeled high-quality datasets for multi-label classification problems, however, is extremely expensive, and sometimes even infeasible, with respect to annotation efforts, especially when the label spaces are too large. This motivates the research on partial-label classification, where only a limited number of labels are annotated and the others are missing. To address this problem, we first propose a pseudo-label based approach to reduce the cost of annotation without bringing additional complexity to the existing classification networks. Then we quantitatively study the impact of missing labels on the performance of classifier. Furthermore, by designing a novel loss function, we are able to relax the requirement that each instance must contain at least one positive label, which is commonly used in most existing approaches. Through comprehensive experiments on three large-scale multi-label image datasets, i.e. MS-COCO, NUS-WIDE, and Pascal VOC12, we show that our method can handle the imbalance between positive labels and negative labels, while still outperforming existing missing-label learning approaches in most cases, and in some cases even approaches with fully labeled datasets.
Probabilistic Generative Transformer Language models for Generative Design of Molecules
Sep 20, 2022
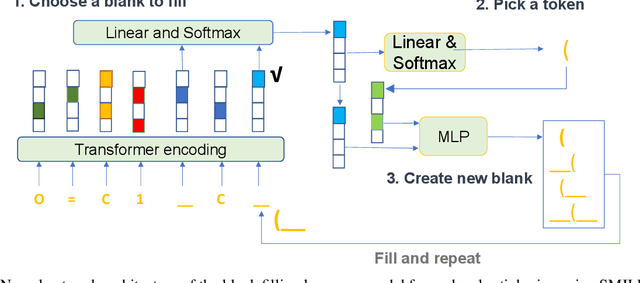
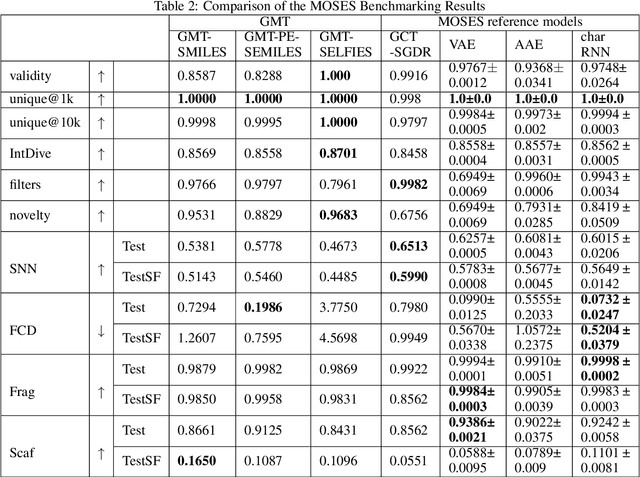
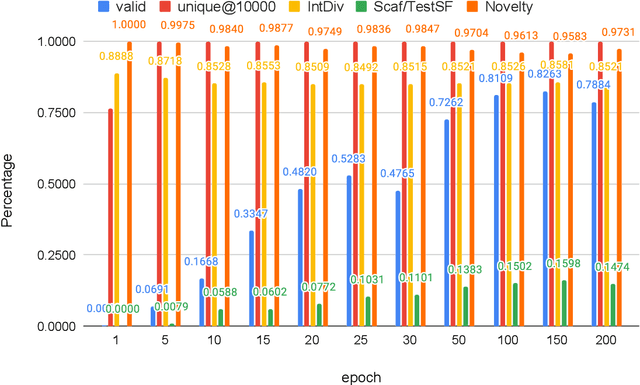
Self-supervised neural language models have recently found wide applications in generative design of organic molecules and protein sequences as well as representation learning for downstream structure classification and functional prediction. However, most of the existing deep learning models for molecule design usually require a big dataset and have a black-box architecture, which makes it difficult to interpret their design logic. Here we propose Generative Molecular Transformer (GMTransformer), a probabilistic neural network model for generative design of molecules. Our model is built on the blank filling language model originally developed for text processing, which has demonstrated unique advantages in learning the "molecules grammars" with high-quality generation, interpretability, and data efficiency. Benchmarked on the MOSES datasets, our models achieve high novelty and Scaf compared to other baselines. The probabilistic generation steps have the potential in tinkering molecule design due to their capability of recommending how to modify existing molecules with explanation, guided by the learned implicit molecule chemistry. The source code and datasets can be accessed freely at https://github.com/usccolumbia/GMTransformer