 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOut-of-distribution materials property prediction using adversarial learning based fine-tuning
Aug 17, 2024



The accurate prediction of material properties is crucial in a wide range of scientific and engineering disciplines. Machine learning (ML) has advanced the state of the art in this field, enabling scientists to discover novel materials and design materials with specific desired properties. However, one major challenge that persists in material property prediction is the generalization of models to out-of-distribution (OOD) samples,i.e., samples that differ significantly from those encountered during training. In this paper, we explore the application of advancements in OOD learning approaches to enhance the robustness and reliability of material property prediction models. We propose and apply the Crystal Adversarial Learning (CAL) algorithm for OOD materials property prediction,which generates synthetic data during training to bias the training towards those samples with high prediction uncertainty. We further propose an adversarial learning based targeting finetuning approach to make the model adapted to a particular OOD dataset, as an alternative to traditional fine-tuning. Our experiments demonstrate the success of our CAL algorithm with its high effectiveness in ML with limited samples which commonly occurs in materials science. Our work represents a promising direction toward better OOD learning and materials property prediction.
Deep Learning-Based Classification of Gamma Photon Interactions in Room-Temperature Semiconductor Radiation Detectors
Nov 01, 2023Photon counting radiation detectors have become an integral part of medical imaging modalities such as Positron Emission Tomography or Computed Tomography. One of the most promising detectors is the wide bandgap room temperature semiconductor detectors, which depends on the interaction gamma/x-ray photons with the detector material involves Compton scattering which leads to multiple interaction photon events (MIPEs) of a single photon. For semiconductor detectors like CdZnTeSe (CZTS), which have a high overlap of detected energies between Compton and photoelectric events, it is nearly impossible to distinguish between Compton scattered events from photoelectric events using conventional readout electronics or signal processing algorithms. Herein, we report a deep learning classifier CoPhNet that distinguishes between Compton scattering and photoelectric interactions of gamma/x-ray photons with CdZnTeSe (CZTS) semiconductor detectors. Our CoPhNet model was trained using simulated data to resemble actual CZTS detector pulses and validated using both simulated and experimental data. These results demonstrated that our CoPhNet model can achieve high classification accuracy over the simulated test set. It also holds its performance robustness under operating parameter shifts such as Signal-Noise-Ratio (SNR) and incident energy. Our work thus laid solid foundation for developing next-generation high energy gamma-rays detectors for better biomedical imaging.
Materials Transformers Language Models for Generative Materials Design: a benchmark study
Jun 27, 2022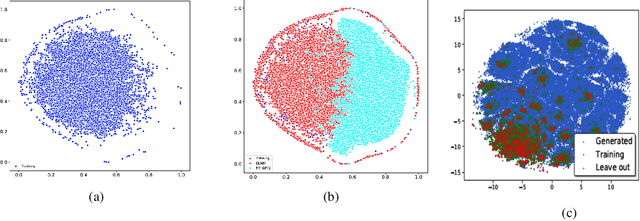
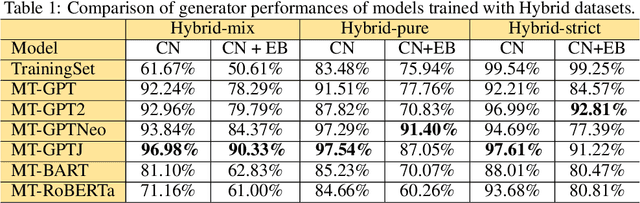
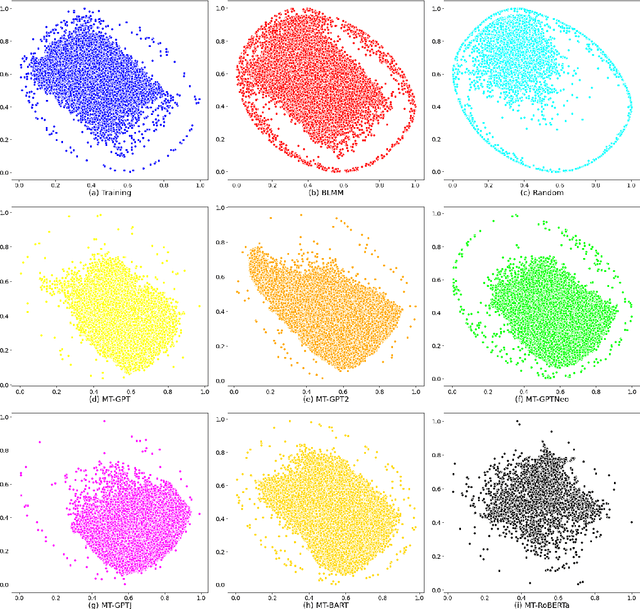
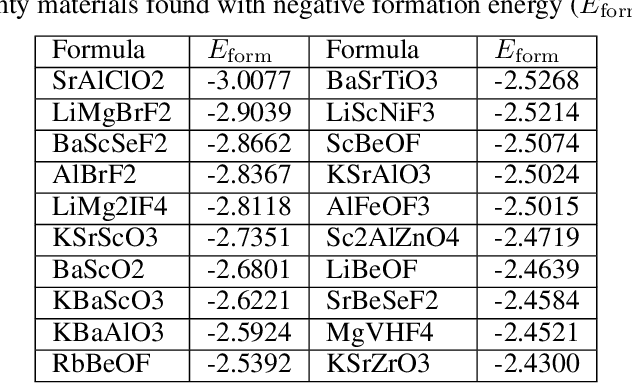
Pre-trained transformer language models on large unlabeled corpus have produced state-of-the-art results in natural language processing, organic molecule design, and protein sequence generation. However, no such models have been applied to learn the composition patterns of inorganic materials. Here we train a series of seven modern transformer language models (GPT, GPT-2, GPT-Neo, GPT-J, BLMM, BART, and RoBERTa) using the expanded formulas from material deposited in the ICSD, OQMD, and Materials Projects databases. Six different datasets with/out non-charge-neutral or balanced electronegativity samples are used to benchmark the performances and uncover the generation biases of modern transformer models for the generative design of materials compositions. Our extensive experiments showed that the causal language models based materials transformers can generate chemically valid materials compositions with as high as 97.54\% to be charge neutral and 91.40\% to be electronegativity balanced, which has more than 6 times higher enrichment compared to a baseline pseudo-random sampling algorithm. These models also demonstrate high novelty and their potential in new materials discovery has been proved by their capability to recover the leave-out materials. We also find that the properties of the generated samples can be tailored by training the models with selected training sets such as high-bandgap materials. Our experiments also showed that different models each have their own preference in terms of the properties of the generated samples and their running time complexity varies a lot. We have applied our materials transformer models to discover a set of new materials as validated using DFT calculations.
Crystal Transformer: Self-learning neural language model for Generative and Tinkering Design of Materials
Apr 25, 2022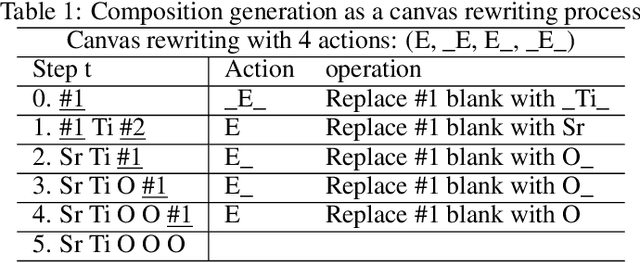
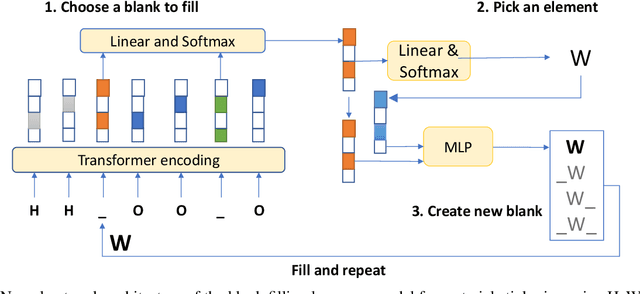
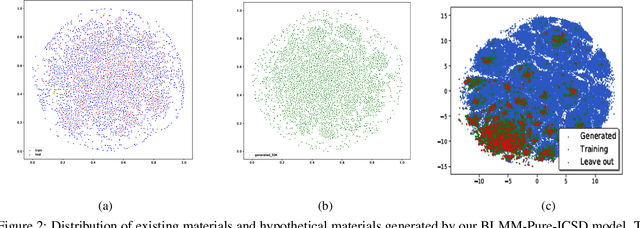
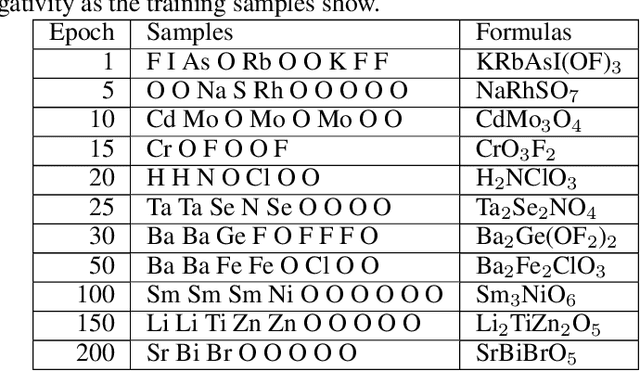
Self-supervised neural language models have recently achieved unprecedented success, from natural language processing to learning the languages of biological sequences and organic molecules. These models have demonstrated superior performance in the generation, structure classification, and functional predictions for proteins and molecules with learned representations. However, most of the masking-based pre-trained language models are not designed for generative design, and their black-box nature makes it difficult to interpret their design logic. Here we propose BLMM Crystal Transformer, a neural network based probabilistic generative model for generative and tinkering design of inorganic materials. Our model is built on the blank filling language model for text generation and has demonstrated unique advantages in learning the "materials grammars" together with high-quality generation, interpretability, and data efficiency. It can generate chemically valid materials compositions with as high as 89.7\% charge neutrality and 84.8\% balanced electronegativity, which are more than 4 and 8 times higher compared to a pseudo random sampling baseline. The probabilistic generation process of BLMM allows it to recommend tinkering operations based on learned materials chemistry and makes it useful for materials doping. Combined with the TCSP crysal structure prediction algorithm, We have applied our model to discover a set of new materials as validated using DFT calculations. Our work thus brings the unsupervised transformer language models based generative artificial intelligence to inorganic materials. A user-friendly web app has been developed for computational materials doping and can be accessed freely at \url{www.materialsatlas.org/blmtinker}.
Scalable deeper graph neural networks for high-performance materials property prediction
Sep 25, 2021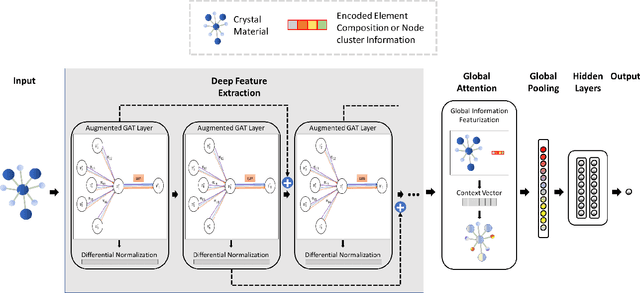
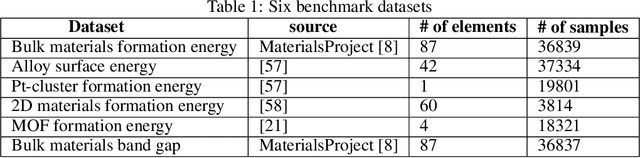
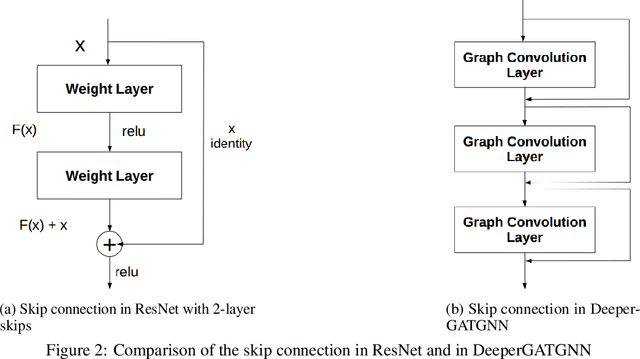
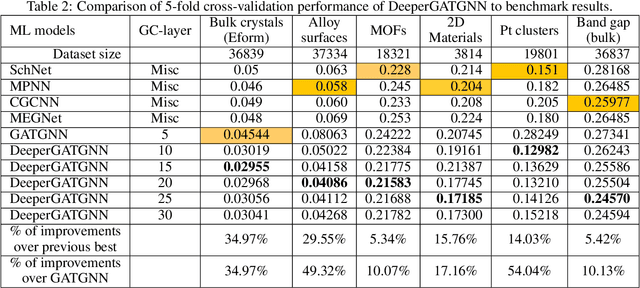
Machine learning (ML) based materials discovery has emerged as one of the most promising approaches for breakthroughs in materials science. While heuristic knowledge based descriptors have been combined with ML algorithms to achieve good performance, the complexity of the physicochemical mechanisms makes it urgently needed to exploit representation learning from either compositions or structures for building highly effective materials machine learning models. Among these methods, the graph neural networks have shown the best performance by its capability to learn high-level features from crystal structures. However, all these models suffer from their inability to scale up the models due to the over-smoothing issue of their message-passing GNN architecture. Here we propose a novel graph attention neural network model DeeperGATGNN with differentiable group normalization and skip-connections, which allows to train very deep graph neural network models (e.g. 30 layers compared to 3-9 layers in previous works). Through systematic benchmark studies over six benchmark datasets for energy and band gap predictions, we show that our scalable DeeperGATGNN model needs little costly hyper-parameter tuning for different datasets and achieves the state-of-the-art prediction performances over five properties out of six with up to 10\% improvement. Our work shows that to deal with the high complexity of mapping the crystal materials structures to their properties, large-scale very deep graph neural networks are needed to achieve robust performances.