 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFacet: highly efficient E(3)-equivariant networks for interatomic potentials
Sep 10, 2025Computational materials discovery is limited by the high cost of first-principles calculations. Machine learning (ML) potentials that predict energies from crystal structures are promising, but existing methods face computational bottlenecks. Steerable graph neural networks (GNNs) encode geometry with spherical harmonics, respecting atomic symmetries -- permutation, rotation, and translation -- for physically realistic predictions. Yet maintaining equivariance is difficult: activation functions must be modified, and each layer must handle multiple data types for different harmonic orders. We present Facet, a GNN architecture for efficient ML potentials, developed through systematic analysis of steerable GNNs. Our innovations include replacing expensive multi-layer perceptrons (MLPs) for interatomic distances with splines, which match performance while cutting computational and memory demands. We also introduce a general-purpose equivariant layer that mixes node information via spherical grid projection followed by standard MLPs -- faster than tensor products and more expressive than linear or gate layers. On the MPTrj dataset, Facet matches leading models with far fewer parameters and under 10% of their training compute. On a crystal relaxation task, it runs twice as fast as MACE models. We further show SevenNet-0's parameters can be reduced by over 25% with no accuracy loss. These techniques enable more than 10x faster training of large-scale foundation models for ML potentials, potentially reshaping computational materials discovery.
Polymorphism Crystal Structure Prediction with Adaptive Space Group Diversity Control
Jun 12, 2025Crystalline materials can form different structural arrangements (i.e. polymorphs) with the same chemical composition, exhibiting distinct physical properties depending on how they were synthesized or the conditions under which they operate. For example, carbon can exist as graphite (soft, conductive) or diamond (hard, insulating). Computational methods that can predict these polymorphs are vital in materials science, which help understand stability relationships, guide synthesis efforts, and discover new materials with desired properties without extensive trial-and-error experimentation. However, effective crystal structure prediction (CSP) algorithms for inorganic polymorph structures remain limited. We propose ParetoCSP2, a multi-objective genetic algorithm for polymorphism CSP that incorporates an adaptive space group diversity control technique, preventing over-representation of any single space group in the population guided by a neural network interatomic potential. Using an improved population initialization method and performing iterative structure relaxation, ParetoCSP2 not only alleviates premature convergence but also achieves improved convergence speed. Our results show that ParetoCSP2 achieves excellent performance in polymorphism prediction, including a nearly perfect space group and structural similarity accuracy for formulas with two polymorphs but with the same number of unit cell atoms. Evaluated on a benchmark dataset, it outperforms baseline algorithms by factors of 2.46-8.62 for these accuracies and improves by 44.8\%-87.04\% across key performance metrics for regular CSP. Our source code is freely available at https://github.com/usccolumbia/ParetoCSP2.
Structure-based out-of-distribution (OOD) materials property prediction: a benchmark study
Jan 16, 2024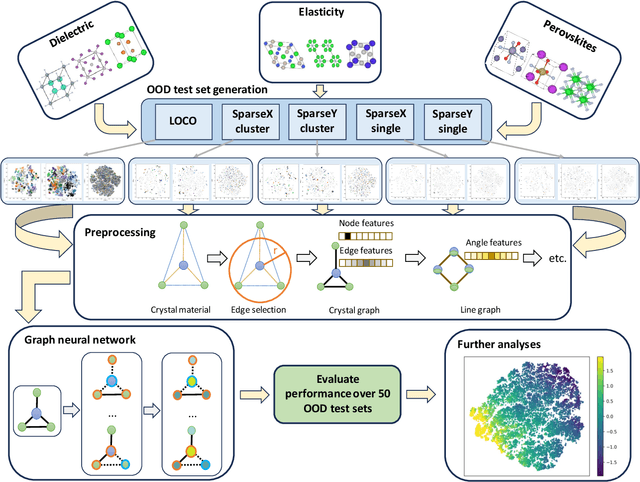
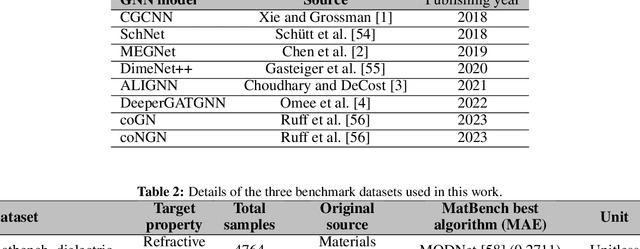
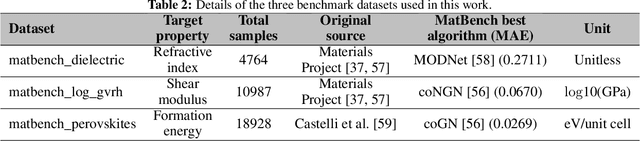
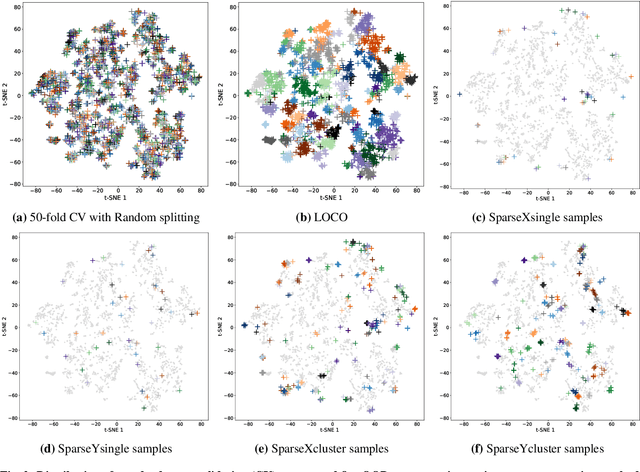
In real-world material research, machine learning (ML) models are usually expected to predict and discover novel exceptional materials that deviate from the known materials. It is thus a pressing question to provide an objective evaluation of ML model performances in property prediction of out-of-distribution (OOD) materials that are different from the training set distribution. Traditional performance evaluation of materials property prediction models through random splitting of the dataset frequently results in artificially high performance assessments due to the inherent redundancy of typical material datasets. Here we present a comprehensive benchmark study of structure-based graph neural networks (GNNs) for extrapolative OOD materials property prediction. We formulate five different categories of OOD ML problems for three benchmark datasets from the MatBench study. Our extensive experiments show that current state-of-the-art GNN algorithms significantly underperform for the OOD property prediction tasks on average compared to their baselines in the MatBench study, demonstrating a crucial generalization gap in realistic material prediction tasks. We further examine the latent physical spaces of these GNN models and identify the sources of CGCNN, ALIGNN, and DeeperGATGNN's significantly more robust OOD performance than those of the current best models in the MatBench study (coGN and coNGN), and provide insights to improve their performance.
Crystal structure prediction using neural network potential and age-fitness Pareto genetic algorithm
Sep 13, 2023While crystal structure prediction (CSP) remains a longstanding challenge, we introduce ParetoCSP, a novel algorithm for CSP, which combines a multi-objective genetic algorithm (MOGA) with a neural network inter-atomic potential (IAP) model to find energetically optimal crystal structures given chemical compositions. We enhance the NSGA-III algorithm by incorporating the genotypic age as an independent optimization criterion and employ the M3GNet universal IAP to guide the GA search. Compared to GN-OA, a state-of-the-art neural potential based CSP algorithm, ParetoCSP demonstrated significantly better predictive capabilities, outperforming by a factor of $2.562$ across $55$ diverse benchmark structures, as evaluated by seven performance metrics. Trajectory analysis of the traversed structures of all algorithms shows that ParetoCSP generated more valid structures than other algorithms, which helped guide the GA to search more effectively for the optimal structures
MD-HIT: Machine learning for materials property prediction with dataset redundancy control
Jul 10, 2023



Materials datasets are usually featured by the existence of many redundant (highly similar) materials due to the tinkering material design practice over the history of materials research. For example, the materials project database has many perovskite cubic structure materials similar to SrTiO$_3$. This sample redundancy within the dataset makes the random splitting of machine learning model evaluation to fail so that the ML models tend to achieve over-estimated predictive performance which is misleading for the materials science community. This issue is well known in the field of bioinformatics for protein function prediction, in which a redundancy reduction procedure (CD-Hit) is always applied to reduce the sample redundancy by ensuring no pair of samples has a sequence similarity greater than a given threshold. This paper surveys the overestimated ML performance in the literature for both composition based and structure based material property prediction. We then propose a material dataset redundancy reduction algorithm called MD-HIT and evaluate it with several composition and structure based distance threshold sfor reducing data set sample redundancy. We show that with this control, the predicted performance tends to better reflect their true prediction capability. Our MD-hit code can be freely accessed at https://github.com/usccolumbia/MD-HIT
Materials Property Prediction with Uncertainty Quantification: A Benchmark Study
Nov 11, 2022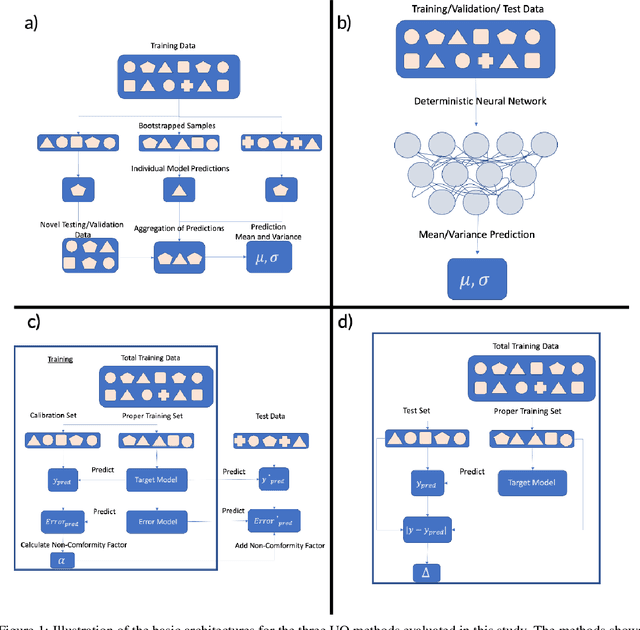
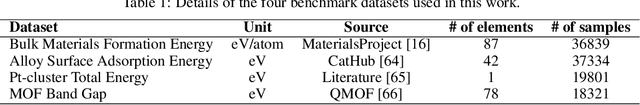
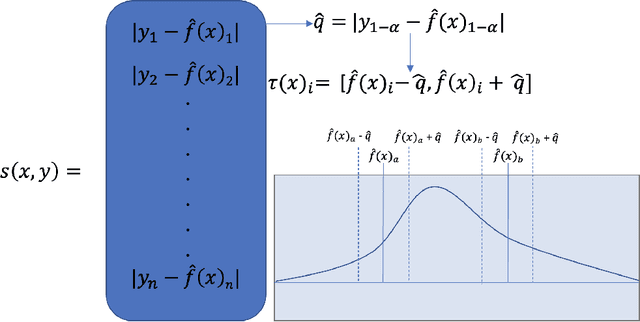
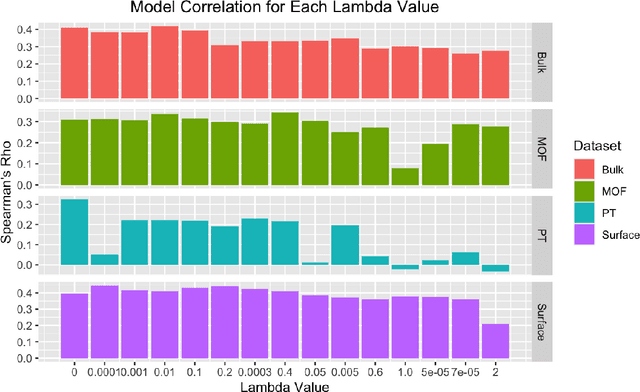
Uncertainty quantification (UQ) has increasing importance in building robust high-performance and generalizable materials property prediction models. It can also be used in active learning to train better models by focusing on getting new training data from uncertain regions. There are several categories of UQ methods each considering different types of uncertainty sources. Here we conduct a comprehensive evaluation on the UQ methods for graph neural network based materials property prediction and evaluate how they truly reflect the uncertainty that we want in error bound estimation or active learning. Our experimental results over four crystal materials datasets (including formation energy, adsorption energy, total energy, and band gap properties) show that the popular ensemble methods for uncertainty estimation is NOT the best choice for UQ in materials property prediction. For the convenience of the community, all the source code and data sets can be accessed freely at \url{https://github.com/usccolumbia/materialsUQ}.
Materials Transformers Language Models for Generative Materials Design: a benchmark study
Jun 27, 2022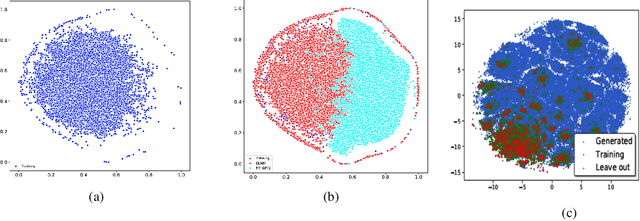
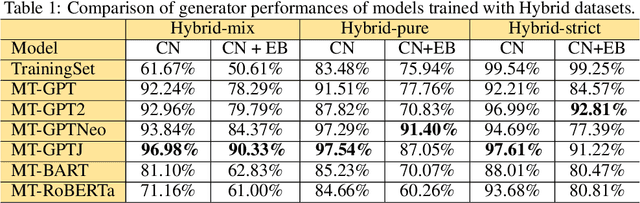
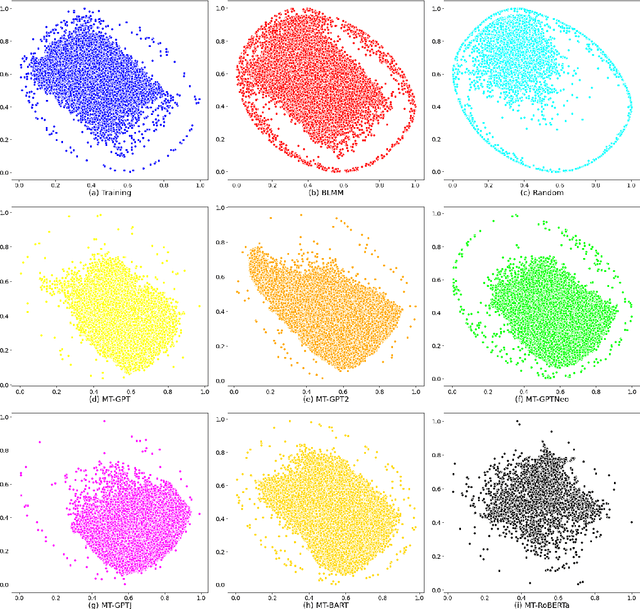
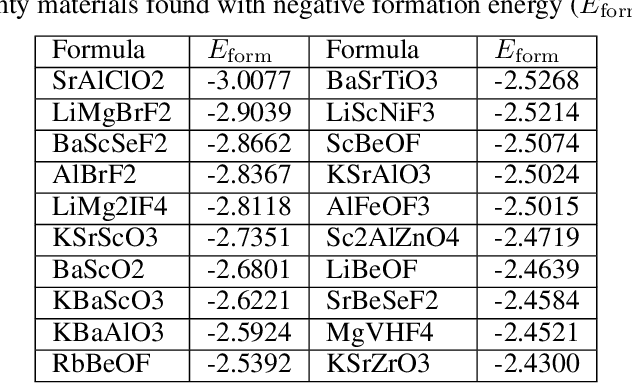
Pre-trained transformer language models on large unlabeled corpus have produced state-of-the-art results in natural language processing, organic molecule design, and protein sequence generation. However, no such models have been applied to learn the composition patterns of inorganic materials. Here we train a series of seven modern transformer language models (GPT, GPT-2, GPT-Neo, GPT-J, BLMM, BART, and RoBERTa) using the expanded formulas from material deposited in the ICSD, OQMD, and Materials Projects databases. Six different datasets with/out non-charge-neutral or balanced electronegativity samples are used to benchmark the performances and uncover the generation biases of modern transformer models for the generative design of materials compositions. Our extensive experiments showed that the causal language models based materials transformers can generate chemically valid materials compositions with as high as 97.54\% to be charge neutral and 91.40\% to be electronegativity balanced, which has more than 6 times higher enrichment compared to a baseline pseudo-random sampling algorithm. These models also demonstrate high novelty and their potential in new materials discovery has been proved by their capability to recover the leave-out materials. We also find that the properties of the generated samples can be tailored by training the models with selected training sets such as high-bandgap materials. Our experiments also showed that different models each have their own preference in terms of the properties of the generated samples and their running time complexity varies a lot. We have applied our materials transformer models to discover a set of new materials as validated using DFT calculations.
Scalable deeper graph neural networks for high-performance materials property prediction
Sep 25, 2021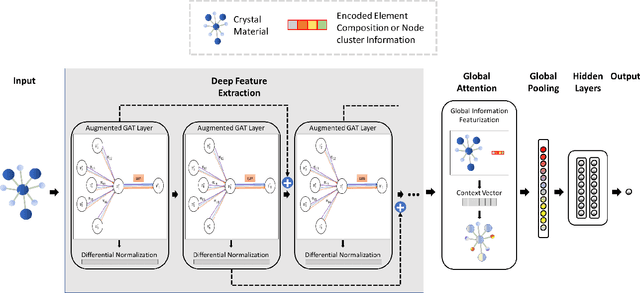
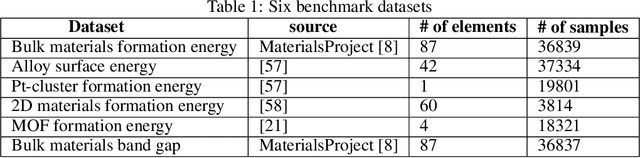
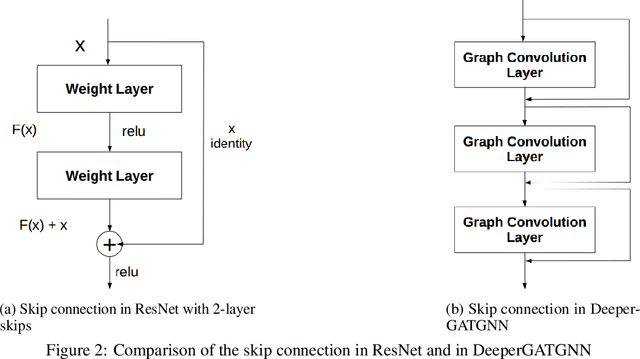
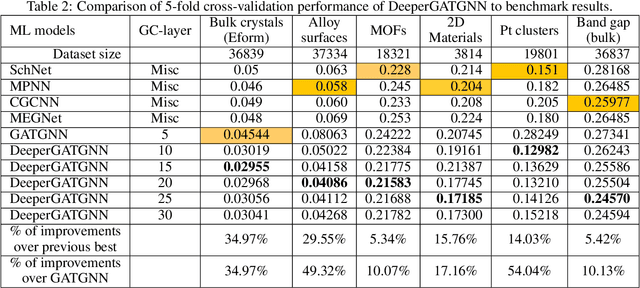
Machine learning (ML) based materials discovery has emerged as one of the most promising approaches for breakthroughs in materials science. While heuristic knowledge based descriptors have been combined with ML algorithms to achieve good performance, the complexity of the physicochemical mechanisms makes it urgently needed to exploit representation learning from either compositions or structures for building highly effective materials machine learning models. Among these methods, the graph neural networks have shown the best performance by its capability to learn high-level features from crystal structures. However, all these models suffer from their inability to scale up the models due to the over-smoothing issue of their message-passing GNN architecture. Here we propose a novel graph attention neural network model DeeperGATGNN with differentiable group normalization and skip-connections, which allows to train very deep graph neural network models (e.g. 30 layers compared to 3-9 layers in previous works). Through systematic benchmark studies over six benchmark datasets for energy and band gap predictions, we show that our scalable DeeperGATGNN model needs little costly hyper-parameter tuning for different datasets and achieves the state-of-the-art prediction performances over five properties out of six with up to 10\% improvement. Our work shows that to deal with the high complexity of mapping the crystal materials structures to their properties, large-scale very deep graph neural networks are needed to achieve robust performances.
MaterialsAtlas.org: A Materials Informatics Web App Platform for Materials Discovery and Survey of State-of-the-Art
Sep 09, 2021
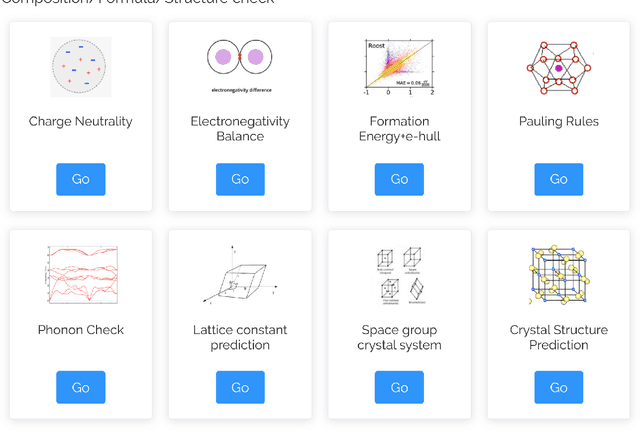
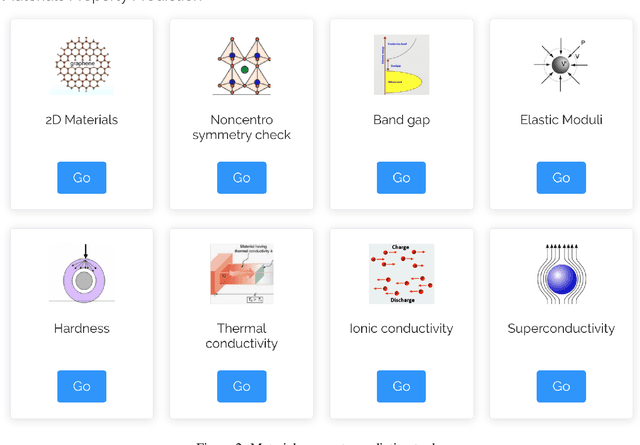
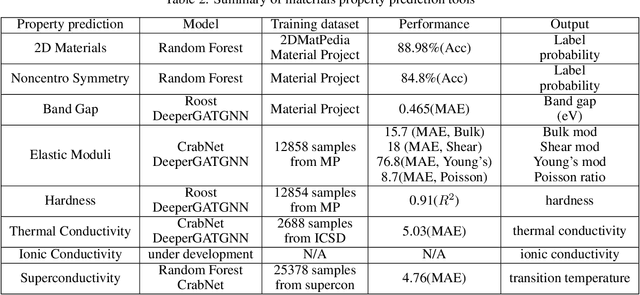
The availability and easy access of large scale experimental and computational materials data have enabled the emergence of accelerated development of algorithms and models for materials property prediction, structure prediction, and generative design of materials. However, lack of user-friendly materials informatics web servers has severely constrained the wide adoption of such tools in the daily practice of materials screening, tinkering, and design space exploration by materials scientists. Herein we first survey current materials informatics web apps and then propose and develop MaterialsAtlas.org, a web based materials informatics toolbox for materials discovery, which includes a variety of routinely needed tools for exploratory materials discovery, including materials composition and structure check (e.g. for neutrality, electronegativity balance, dynamic stability, Pauling rules), materials property prediction (e.g. band gap, elastic moduli, hardness, thermal conductivity), and search for hypothetical materials. These user-friendly tools can be freely accessed at \url{www.materialsatlas.org}. We argue that such materials informatics apps should be widely developed by the community to speed up the materials discovery processes.