 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFacet: highly efficient E(3)-equivariant networks for interatomic potentials
Sep 10, 2025Computational materials discovery is limited by the high cost of first-principles calculations. Machine learning (ML) potentials that predict energies from crystal structures are promising, but existing methods face computational bottlenecks. Steerable graph neural networks (GNNs) encode geometry with spherical harmonics, respecting atomic symmetries -- permutation, rotation, and translation -- for physically realistic predictions. Yet maintaining equivariance is difficult: activation functions must be modified, and each layer must handle multiple data types for different harmonic orders. We present Facet, a GNN architecture for efficient ML potentials, developed through systematic analysis of steerable GNNs. Our innovations include replacing expensive multi-layer perceptrons (MLPs) for interatomic distances with splines, which match performance while cutting computational and memory demands. We also introduce a general-purpose equivariant layer that mixes node information via spherical grid projection followed by standard MLPs -- faster than tensor products and more expressive than linear or gate layers. On the MPTrj dataset, Facet matches leading models with far fewer parameters and under 10% of their training compute. On a crystal relaxation task, it runs twice as fast as MACE models. We further show SevenNet-0's parameters can be reduced by over 25% with no accuracy loss. These techniques enable more than 10x faster training of large-scale foundation models for ML potentials, potentially reshaping computational materials discovery.
Polymorphism Crystal Structure Prediction with Adaptive Space Group Diversity Control
Jun 12, 2025Crystalline materials can form different structural arrangements (i.e. polymorphs) with the same chemical composition, exhibiting distinct physical properties depending on how they were synthesized or the conditions under which they operate. For example, carbon can exist as graphite (soft, conductive) or diamond (hard, insulating). Computational methods that can predict these polymorphs are vital in materials science, which help understand stability relationships, guide synthesis efforts, and discover new materials with desired properties without extensive trial-and-error experimentation. However, effective crystal structure prediction (CSP) algorithms for inorganic polymorph structures remain limited. We propose ParetoCSP2, a multi-objective genetic algorithm for polymorphism CSP that incorporates an adaptive space group diversity control technique, preventing over-representation of any single space group in the population guided by a neural network interatomic potential. Using an improved population initialization method and performing iterative structure relaxation, ParetoCSP2 not only alleviates premature convergence but also achieves improved convergence speed. Our results show that ParetoCSP2 achieves excellent performance in polymorphism prediction, including a nearly perfect space group and structural similarity accuracy for formulas with two polymorphs but with the same number of unit cell atoms. Evaluated on a benchmark dataset, it outperforms baseline algorithms by factors of 2.46-8.62 for these accuracies and improves by 44.8\%-87.04\% across key performance metrics for regular CSP. Our source code is freely available at https://github.com/usccolumbia/ParetoCSP2.
Scalable deeper graph neural networks for high-performance materials property prediction
Sep 25, 2021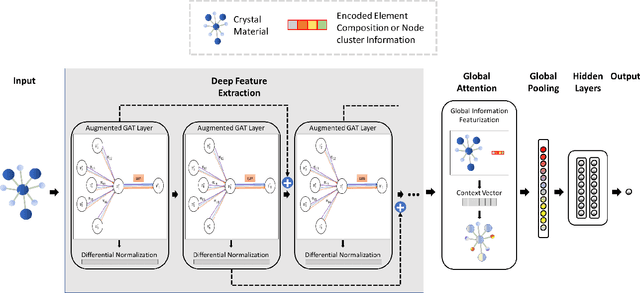
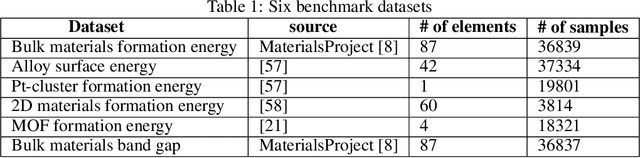
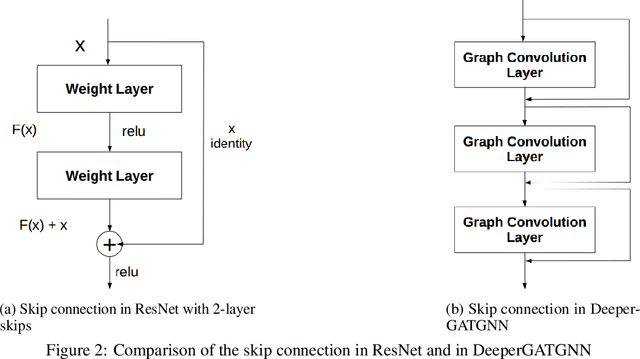
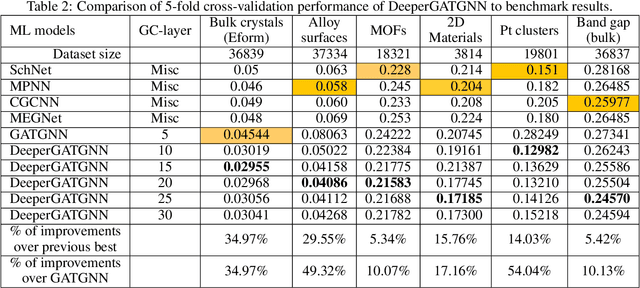
Machine learning (ML) based materials discovery has emerged as one of the most promising approaches for breakthroughs in materials science. While heuristic knowledge based descriptors have been combined with ML algorithms to achieve good performance, the complexity of the physicochemical mechanisms makes it urgently needed to exploit representation learning from either compositions or structures for building highly effective materials machine learning models. Among these methods, the graph neural networks have shown the best performance by its capability to learn high-level features from crystal structures. However, all these models suffer from their inability to scale up the models due to the over-smoothing issue of their message-passing GNN architecture. Here we propose a novel graph attention neural network model DeeperGATGNN with differentiable group normalization and skip-connections, which allows to train very deep graph neural network models (e.g. 30 layers compared to 3-9 layers in previous works). Through systematic benchmark studies over six benchmark datasets for energy and band gap predictions, we show that our scalable DeeperGATGNN model needs little costly hyper-parameter tuning for different datasets and achieves the state-of-the-art prediction performances over five properties out of six with up to 10\% improvement. Our work shows that to deal with the high complexity of mapping the crystal materials structures to their properties, large-scale very deep graph neural networks are needed to achieve robust performances.
Modeling and Optimizing Laser-Induced Graphene
Jul 29, 2021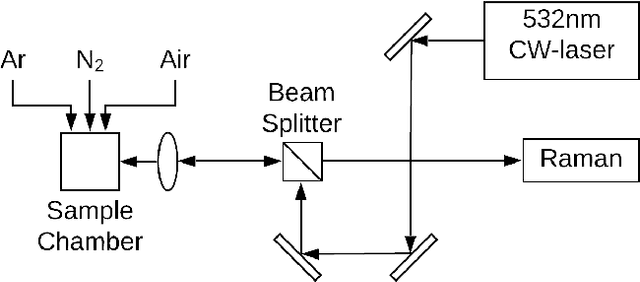
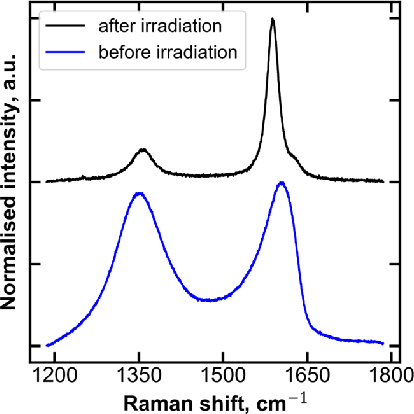
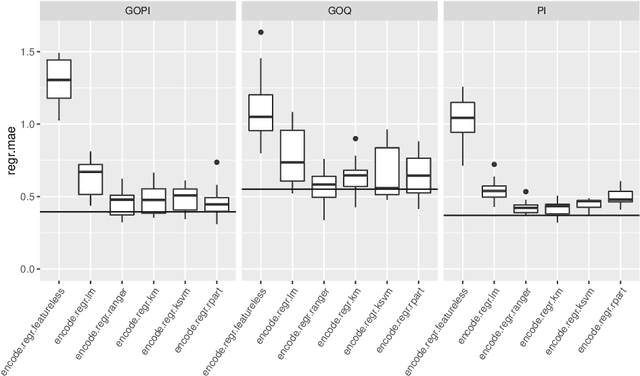
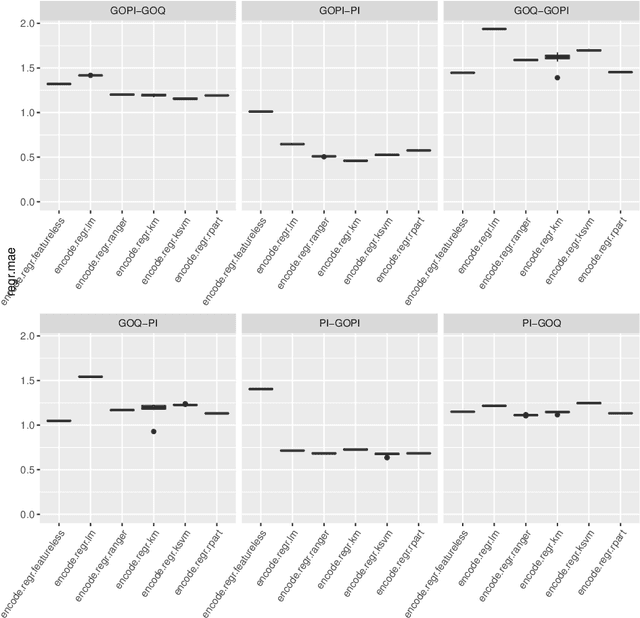
A lot of technological advances depend on next-generation materials, such as graphene, which enables a raft of new applications, for example better electronics. Manufacturing such materials is often difficult; in particular, producing graphene at scale is an open problem. We provide a series of datasets that describe the optimization of the production of laser-induced graphene, an established manufacturing method that has shown great promise. We pose three challenges based on the datasets we provide -- modeling the behavior of laser-induced graphene production with respect to parameters of the production process, transferring models and knowledge between different precursor materials, and optimizing the outcome of the transformation over the space of possible production parameters. We present illustrative results, along with the code used to generate them, as a starting point for interested users. The data we provide represents an important real-world application of machine learning; to the best of our knowledge, no similar datasets are available.