 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe Data Fusion Labeler (dFL): Challenges and Solutions to Data Harmonization, Labeling, and Provenance in Fusion Energy
Nov 12, 2025Fusion energy research increasingly depends on the ability to integrate heterogeneous, multimodal datasets from high-resolution diagnostics, control systems, and multiscale simulations. The sheer volume and complexity of these datasets demand the development of new tools capable of systematically harmonizing and extracting knowledge across diverse modalities. The Data Fusion Labeler (dFL) is introduced as a unified workflow instrument that performs uncertainty-aware data harmonization, schema-compliant data fusion, and provenance-rich manual and automated labeling at scale. By embedding alignment, normalization, and labeling within a reproducible, operator-order-aware framework, dFL reduces time-to-analysis by greater than 50X (e.g., enabling >200 shots/hour to be consistently labeled rather than a handful per day), enhances label (and subsequently training) quality, and enables cross-device comparability. Case studies from DIII-D demonstrate its application to automated ELM detection and confinement regime classification, illustrating its potential as a core component of data-driven discovery, model validation, and real-time control in future burning plasma devices.
Predicting Lattice Phonon Vibrational Frequencies Using Deep Graph Neural Networks
Nov 10, 2021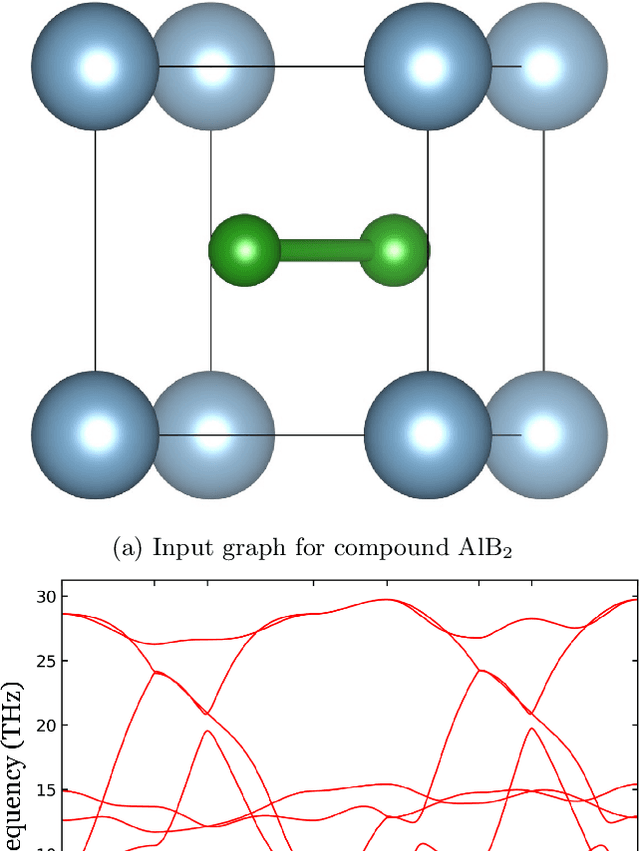
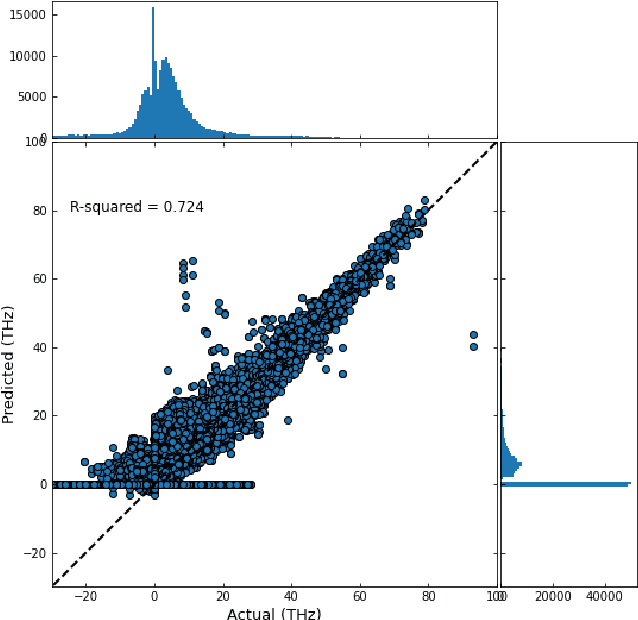
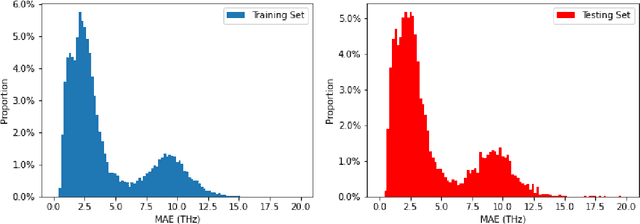
Lattice vibration frequencies are related to many important materials properties such as thermal and electrical conductivity as well as superconductivity. However, computational calculation of vibration frequencies using density functional theory (DFT) methods is too computationally demanding for a large number of samples in materials screening. Here we propose a deep graph neural network-based algorithm for predicting crystal vibration frequencies from crystal structures with high accuracy. Our algorithm addresses the variable dimension of vibration frequency spectrum using the zero padding scheme. Benchmark studies on two data sets with 15,000 and 35,552 samples show that the aggregated $R^2$ scores of the prediction reaches 0.554 and 0.724 respectively. Our work demonstrates the capability of deep graph neural networks to learn to predict phonon spectrum properties of crystal structures in addition to phonon density of states (DOS) and electronic DOS in which the output dimension is constant.
Scalable deeper graph neural networks for high-performance materials property prediction
Sep 25, 2021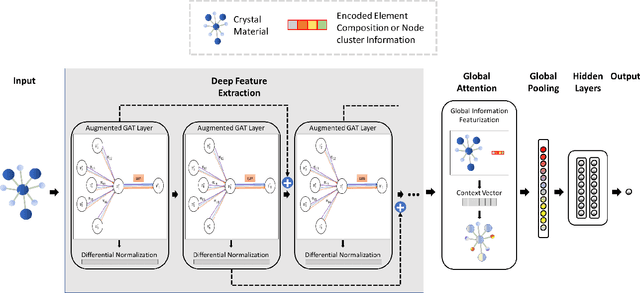
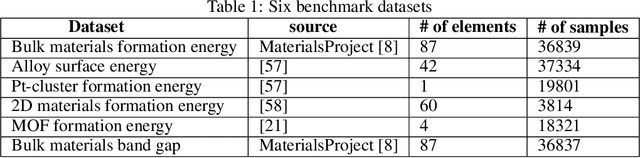
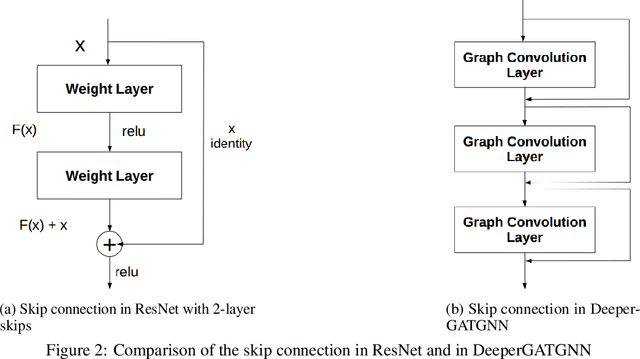
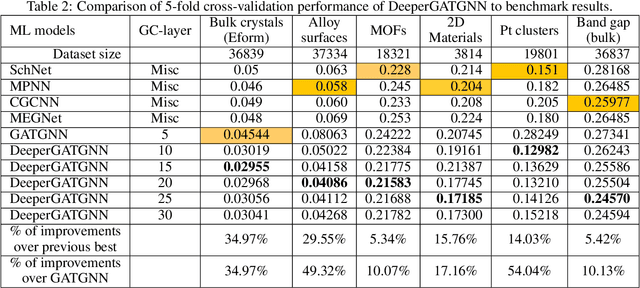
Machine learning (ML) based materials discovery has emerged as one of the most promising approaches for breakthroughs in materials science. While heuristic knowledge based descriptors have been combined with ML algorithms to achieve good performance, the complexity of the physicochemical mechanisms makes it urgently needed to exploit representation learning from either compositions or structures for building highly effective materials machine learning models. Among these methods, the graph neural networks have shown the best performance by its capability to learn high-level features from crystal structures. However, all these models suffer from their inability to scale up the models due to the over-smoothing issue of their message-passing GNN architecture. Here we propose a novel graph attention neural network model DeeperGATGNN with differentiable group normalization and skip-connections, which allows to train very deep graph neural network models (e.g. 30 layers compared to 3-9 layers in previous works). Through systematic benchmark studies over six benchmark datasets for energy and band gap predictions, we show that our scalable DeeperGATGNN model needs little costly hyper-parameter tuning for different datasets and achieves the state-of-the-art prediction performances over five properties out of six with up to 10\% improvement. Our work shows that to deal with the high complexity of mapping the crystal materials structures to their properties, large-scale very deep graph neural networks are needed to achieve robust performances.
MaterialsAtlas.org: A Materials Informatics Web App Platform for Materials Discovery and Survey of State-of-the-Art
Sep 09, 2021
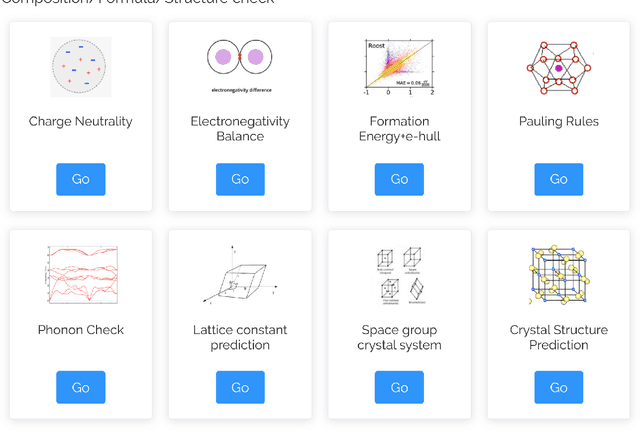
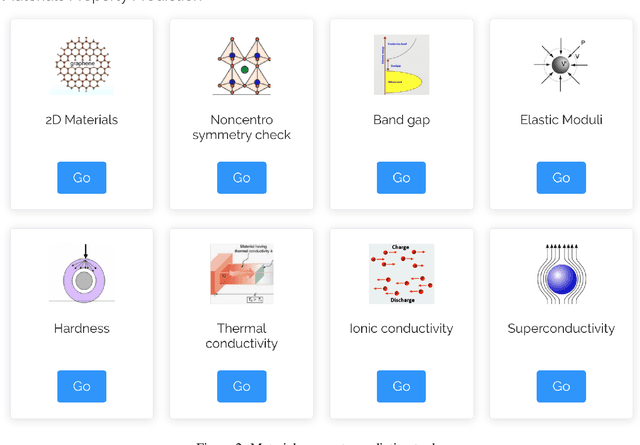
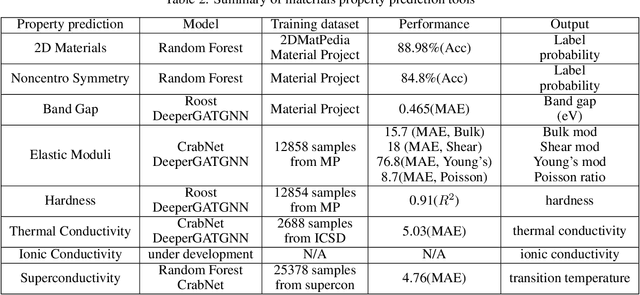
The availability and easy access of large scale experimental and computational materials data have enabled the emergence of accelerated development of algorithms and models for materials property prediction, structure prediction, and generative design of materials. However, lack of user-friendly materials informatics web servers has severely constrained the wide adoption of such tools in the daily practice of materials screening, tinkering, and design space exploration by materials scientists. Herein we first survey current materials informatics web apps and then propose and develop MaterialsAtlas.org, a web based materials informatics toolbox for materials discovery, which includes a variety of routinely needed tools for exploratory materials discovery, including materials composition and structure check (e.g. for neutrality, electronegativity balance, dynamic stability, Pauling rules), materials property prediction (e.g. band gap, elastic moduli, hardness, thermal conductivity), and search for hypothetical materials. These user-friendly tools can be freely accessed at \url{www.materialsatlas.org}. We argue that such materials informatics apps should be widely developed by the community to speed up the materials discovery processes.
Active learning based generative design for the discovery of wide bandgap materials
Feb 28, 2021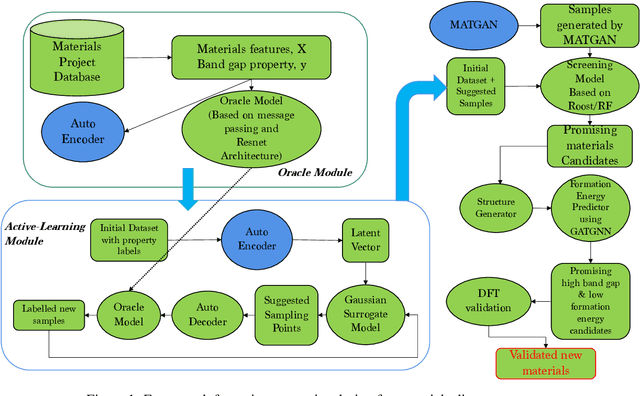
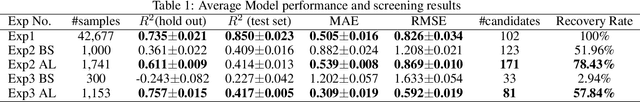
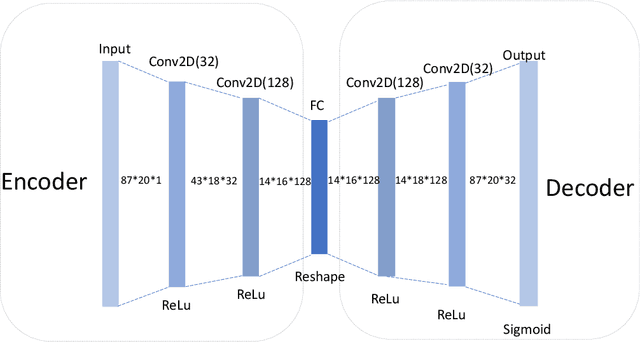
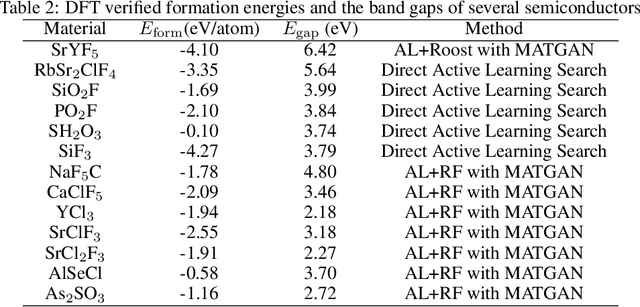
Active learning has been increasingly applied to screening functional materials from existing materials databases with desired properties. However, the number of known materials deposited in the popular materials databases such as ICSD and Materials Project is extremely limited and consists of just a tiny portion of the vast chemical design space. Herein we present an active generative inverse design method that combines active learning with a deep variational autoencoder neural network and a generative adversarial deep neural network model to discover new materials with a target property in the whole chemical design space. The application of this method has allowed us to discover new thermodynamically stable materials with high band gap (SrYF$_5$) and semiconductors with specified band gap ranges (SrClF$_3$, CaClF$_5$, YCl$_3$, SrC$_2$F$_3$, AlSCl, As$_2$O$_3$), all of which are verified by the first principle DFT calculations. Our experiments show that while active learning itself may sample chemically infeasible candidates, these samples help to train effective screening models for filtering out materials with desired properties from the hypothetical materials created by the generative model. The experiments show the effectiveness of our active generative inverse design approach.
NODE-SELECT: A Graph Neural Network Based On A Selective Propagation Technique
Feb 17, 2021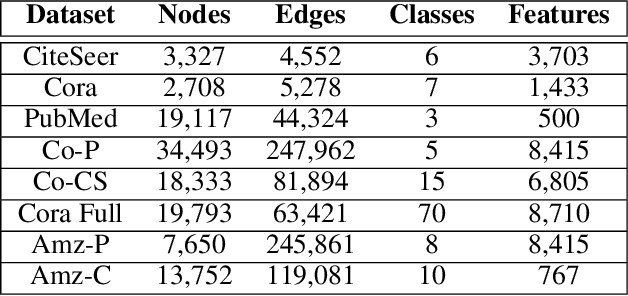
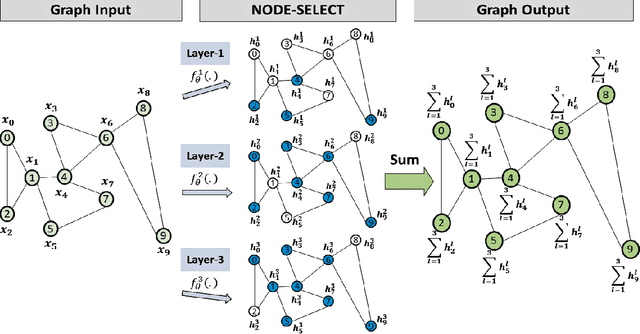
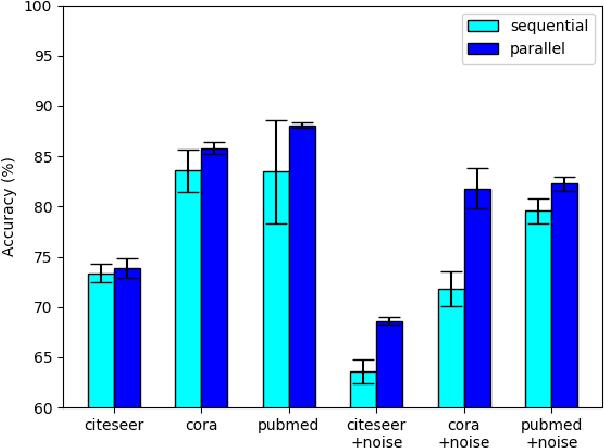
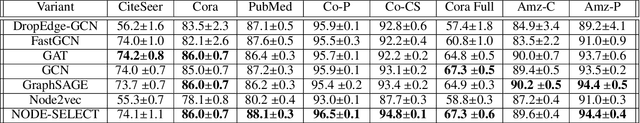
While there exists a wide variety of graph neural networks (GNN) for node classification, only a minority of them adopt mechanisms that effectively target noise propagation during the message-passing procedure. Additionally, a very important challenge that significantly affects graph neural networks is the issue of scalability which limits their application to larger graphs. In this paper we propose our method named NODE-SELECT: an efficient graph neural network that uses subsetting layers which only allow the best sharing-fitting nodes to propagate their information. By having a selection mechanism within each layer which we stack in parallel, our proposed method NODE-SELECT is able to both reduce the amount noise propagated and adapt the restrictive sharing concept observed in real world graphs. Our NODE-SELECT significantly outperformed existing GNN frameworks in noise experiments and matched state-of-the art results in experiments without noise over different benchmark datasets.
Predicting Elastic Properties of Materials from Electronic Charge Density Using 3D Deep Convolutional Neural Networks
Apr 11, 2020
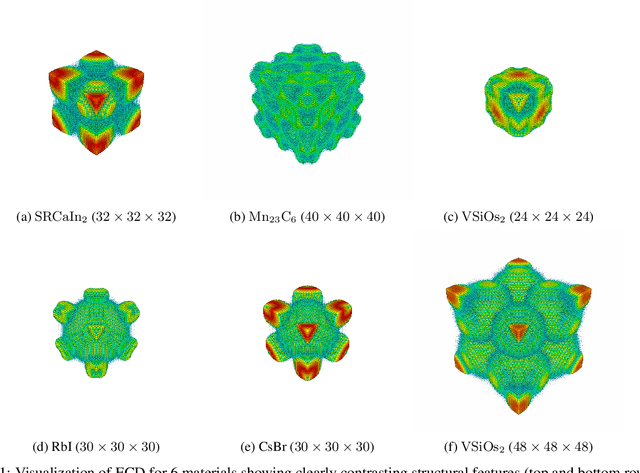
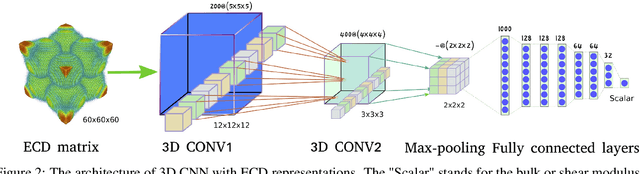

Materials representation plays a key role in machine learning based prediction of materials properties and new materials discovery. Currently both graph and 3D voxel representation methods are based on the heterogeneous elements of the crystal structures. Here, we propose to use electronic charge density (ECD) as a generic unified 3D descriptor for materials property prediction with the advantage of possessing close relation with the physical and chemical properties of materials. We developed an ECD based 3D convolutional neural networks (CNNs) for predicting elastic properties of materials, in which CNNs can learn effective hierarchical features with multiple convolving and pooling operations. Extensive benchmark experiments over 2,170 Fm-3m face-centered-cubic (FCC) materials show that our ECD based CNNs can achieve good performance for elasticity prediction. Especially, our CNN models based on the fusion of elemental Magpie features and ECD descriptors achieved the best 5-fold cross-validation performance. More importantly, we showed that our ECD based CNN models can achieve significantly better extrapolation performance when evaluated over non-redundant datasets where there are few neighbor training samples around test samples. As additional validation, we evaluated the predictive performance of our models on 329 materials of space group Fm-3m by comparing to DFT calculated values, which shows better prediction power of our model for bulk modulus than shear modulus. Due to the unified representation power of ECD, it is expected that our ECD based CNN approach can also be applied to predict other physical and chemical properties of crystalline materials.
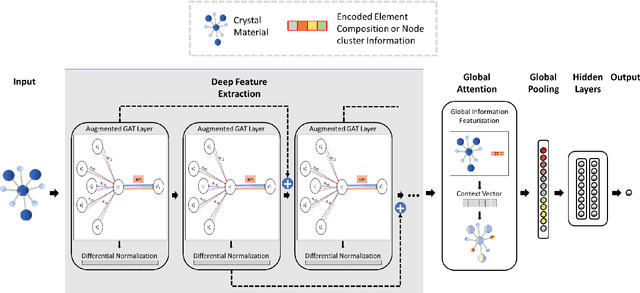
Global Attention based Graph Convolutional Neural Networks for Improved Materials Property Prediction
Mar 11, 2020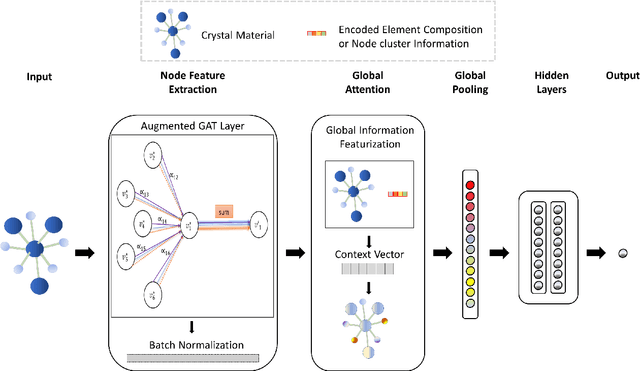

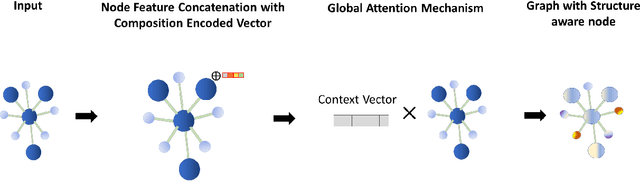
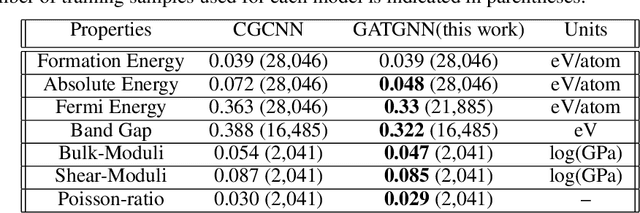
Machine learning (ML) methods have gained increasing popularity in exploring and developing new materials. More specifically, graph neural network (GNN) has been applied in predicting material properties. In this work, we develop a novel model, GATGNN, for predicting inorganic material properties based on graph neural networks composed of multiple graph-attention layers (GAT) and a global attention layer. Through the application of the GAT layers, our model can efficiently learn the complex bonds shared among the atoms within each atom's local neighborhood. Subsequently, the global attention layer provides the weight coefficients of each atom in the inorganic crystal material which are used to considerably improve our model's performance. Notably, with the development of our GATGNN model, we show that our method is able to both outperform the previous models' predictions and provide insight into the crystallization of the material.