 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFoundation-Model Surrogates Enable Data-Efficient Active Learning for Materials Discovery
Mar 17, 2026Active learning (AL) has emerged as a powerful paradigm for accelerating materials discovery by iteratively steering experiments toward promising candidates, reducing the number of costly synthesis-and-characterization cycles needed to identify optimal materials. However, current AL relies predominantly on Gaussian Process (GP) and Random Forest (RF) surrogates, which suffer from complementary limitations: GP underfits complex composition-property landscapes due to rigid kernel assumptions, while RF produces unreliable heuristic uncertainty estimates in small-data regimes. This small-data challenge is pervasive in materials science, making reliable surrogate modeling extremely difficult with models trained from scratch on each new dataset. Here we propose In-Context Active Learning (ICAL), which addresses this bottleneck by replacing conventional surrogates with TabPFN, a transformer-based foundation model (FM) pre-trained on millions of synthetic regression tasks to meta-learn a universal prior over tabular data, upon which TabPFN performs principled Bayesian inference in a single forward pass without dataset-specific retraining, delivering strong small-data regression performance and well-calibrated predictive uncertainty (required for effective AL). We benchmark ICAL against GP and RF across 10 materials datasets and TabPFN wins on 8 out of 10 datasets, achieving a mean saving of 52% in extra evaluations relative to GP and 29.77% relative to RF. Cross-validation analysis confirms that TabPFN's advantage stems from superior uncertainty calibration, achieving the lowest Negative Log-Likelihood and Area Under the Sparsification Error curve among all surrogates. These results demonstrate that pre-trained FMs can serve as effective surrogates for active learning, enabling data-efficient discovery across diverse materials systems and small-data experimental sciences.
Facet: highly efficient E(3)-equivariant networks for interatomic potentials
Sep 10, 2025Computational materials discovery is limited by the high cost of first-principles calculations. Machine learning (ML) potentials that predict energies from crystal structures are promising, but existing methods face computational bottlenecks. Steerable graph neural networks (GNNs) encode geometry with spherical harmonics, respecting atomic symmetries -- permutation, rotation, and translation -- for physically realistic predictions. Yet maintaining equivariance is difficult: activation functions must be modified, and each layer must handle multiple data types for different harmonic orders. We present Facet, a GNN architecture for efficient ML potentials, developed through systematic analysis of steerable GNNs. Our innovations include replacing expensive multi-layer perceptrons (MLPs) for interatomic distances with splines, which match performance while cutting computational and memory demands. We also introduce a general-purpose equivariant layer that mixes node information via spherical grid projection followed by standard MLPs -- faster than tensor products and more expressive than linear or gate layers. On the MPTrj dataset, Facet matches leading models with far fewer parameters and under 10% of their training compute. On a crystal relaxation task, it runs twice as fast as MACE models. We further show SevenNet-0's parameters can be reduced by over 25% with no accuracy loss. These techniques enable more than 10x faster training of large-scale foundation models for ML potentials, potentially reshaping computational materials discovery.
Polymorphism Crystal Structure Prediction with Adaptive Space Group Diversity Control
Jun 12, 2025Crystalline materials can form different structural arrangements (i.e. polymorphs) with the same chemical composition, exhibiting distinct physical properties depending on how they were synthesized or the conditions under which they operate. For example, carbon can exist as graphite (soft, conductive) or diamond (hard, insulating). Computational methods that can predict these polymorphs are vital in materials science, which help understand stability relationships, guide synthesis efforts, and discover new materials with desired properties without extensive trial-and-error experimentation. However, effective crystal structure prediction (CSP) algorithms for inorganic polymorph structures remain limited. We propose ParetoCSP2, a multi-objective genetic algorithm for polymorphism CSP that incorporates an adaptive space group diversity control technique, preventing over-representation of any single space group in the population guided by a neural network interatomic potential. Using an improved population initialization method and performing iterative structure relaxation, ParetoCSP2 not only alleviates premature convergence but also achieves improved convergence speed. Our results show that ParetoCSP2 achieves excellent performance in polymorphism prediction, including a nearly perfect space group and structural similarity accuracy for formulas with two polymorphs but with the same number of unit cell atoms. Evaluated on a benchmark dataset, it outperforms baseline algorithms by factors of 2.46-8.62 for these accuracies and improves by 44.8\%-87.04\% across key performance metrics for regular CSP. Our source code is freely available at https://github.com/usccolumbia/ParetoCSP2.
Out-of-distribution materials property prediction using adversarial learning based fine-tuning
Aug 17, 2024



The accurate prediction of material properties is crucial in a wide range of scientific and engineering disciplines. Machine learning (ML) has advanced the state of the art in this field, enabling scientists to discover novel materials and design materials with specific desired properties. However, one major challenge that persists in material property prediction is the generalization of models to out-of-distribution (OOD) samples,i.e., samples that differ significantly from those encountered during training. In this paper, we explore the application of advancements in OOD learning approaches to enhance the robustness and reliability of material property prediction models. We propose and apply the Crystal Adversarial Learning (CAL) algorithm for OOD materials property prediction,which generates synthetic data during training to bias the training towards those samples with high prediction uncertainty. We further propose an adversarial learning based targeting finetuning approach to make the model adapted to a particular OOD dataset, as an alternative to traditional fine-tuning. Our experiments demonstrate the success of our CAL algorithm with its high effectiveness in ML with limited samples which commonly occurs in materials science. Our work represents a promising direction toward better OOD learning and materials property prediction.
AlphaCrystal-II: Distance matrix based crystal structure prediction using deep learning
Apr 07, 2024



Computational prediction of stable crystal structures has a profound impact on the large-scale discovery of novel functional materials. However, predicting the crystal structure solely from a material's composition or formula is a promising yet challenging task, as traditional ab initio crystal structure prediction (CSP) methods rely on time-consuming global searches and first-principles free energy calculations. Inspired by the recent success of deep learning approaches in protein structure prediction, which utilize pairwise amino acid interactions to describe 3D structures, we present AlphaCrystal-II, a novel knowledge-based solution that exploits the abundant inter-atomic interaction patterns found in existing known crystal structures. AlphaCrystal-II predicts the atomic distance matrix of a target crystal material and employs this matrix to reconstruct its 3D crystal structure. By leveraging the wealth of inter-atomic relationships of known crystal structures, our approach demonstrates remarkable effectiveness and reliability in structure prediction through comprehensive experiments. This work highlights the potential of data-driven methods in accelerating the discovery and design of new materials with tailored properties.
Structure-based out-of-distribution (OOD) materials property prediction: a benchmark study
Jan 16, 2024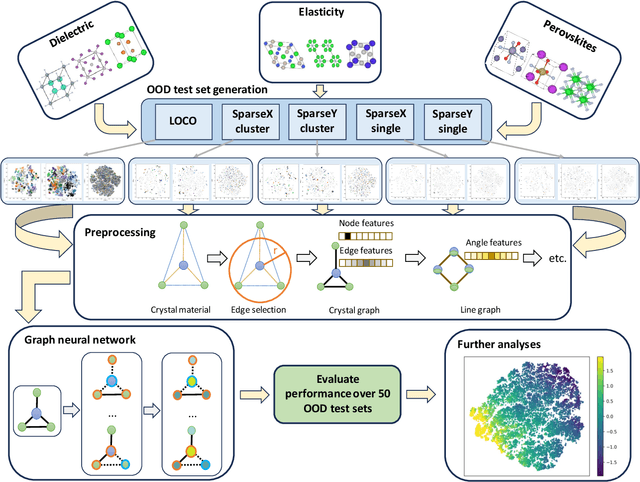
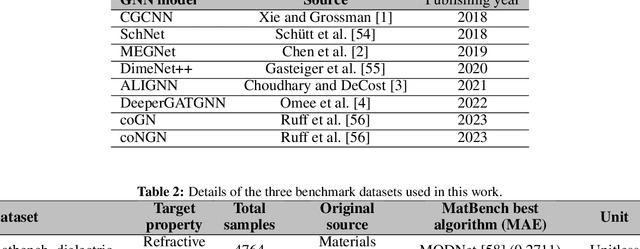
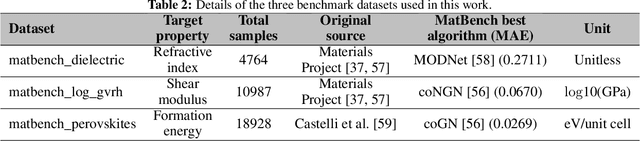
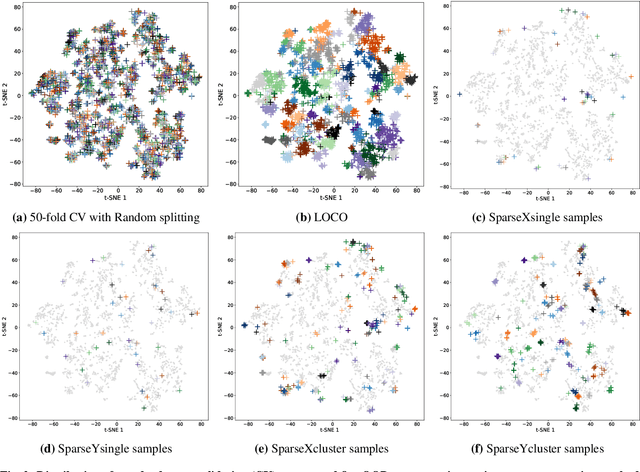
In real-world material research, machine learning (ML) models are usually expected to predict and discover novel exceptional materials that deviate from the known materials. It is thus a pressing question to provide an objective evaluation of ML model performances in property prediction of out-of-distribution (OOD) materials that are different from the training set distribution. Traditional performance evaluation of materials property prediction models through random splitting of the dataset frequently results in artificially high performance assessments due to the inherent redundancy of typical material datasets. Here we present a comprehensive benchmark study of structure-based graph neural networks (GNNs) for extrapolative OOD materials property prediction. We formulate five different categories of OOD ML problems for three benchmark datasets from the MatBench study. Our extensive experiments show that current state-of-the-art GNN algorithms significantly underperform for the OOD property prediction tasks on average compared to their baselines in the MatBench study, demonstrating a crucial generalization gap in realistic material prediction tasks. We further examine the latent physical spaces of these GNN models and identify the sources of CGCNN, ALIGNN, and DeeperGATGNN's significantly more robust OOD performance than those of the current best models in the MatBench study (coGN and coNGN), and provide insights to improve their performance.
Deep Learning-Based Classification of Gamma Photon Interactions in Room-Temperature Semiconductor Radiation Detectors
Nov 01, 2023Photon counting radiation detectors have become an integral part of medical imaging modalities such as Positron Emission Tomography or Computed Tomography. One of the most promising detectors is the wide bandgap room temperature semiconductor detectors, which depends on the interaction gamma/x-ray photons with the detector material involves Compton scattering which leads to multiple interaction photon events (MIPEs) of a single photon. For semiconductor detectors like CdZnTeSe (CZTS), which have a high overlap of detected energies between Compton and photoelectric events, it is nearly impossible to distinguish between Compton scattered events from photoelectric events using conventional readout electronics or signal processing algorithms. Herein, we report a deep learning classifier CoPhNet that distinguishes between Compton scattering and photoelectric interactions of gamma/x-ray photons with CdZnTeSe (CZTS) semiconductor detectors. Our CoPhNet model was trained using simulated data to resemble actual CZTS detector pulses and validated using both simulated and experimental data. These results demonstrated that our CoPhNet model can achieve high classification accuracy over the simulated test set. It also holds its performance robustness under operating parameter shifts such as Signal-Noise-Ratio (SNR) and incident energy. Our work thus laid solid foundation for developing next-generation high energy gamma-rays detectors for better biomedical imaging.
Generative Design of inorganic compounds using deep diffusion language models
Sep 30, 2023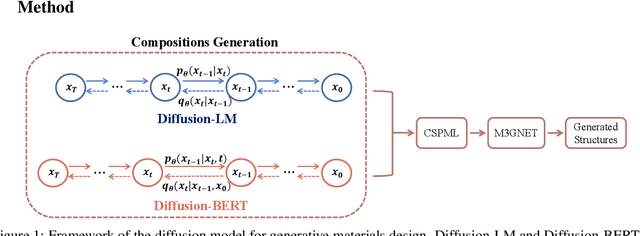
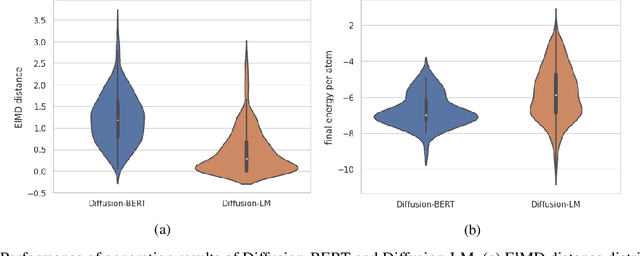
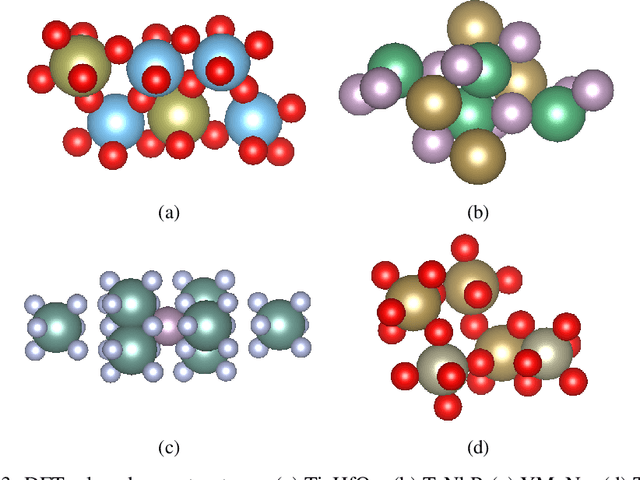
Due to the vast chemical space, discovering materials with a specific function is challenging. Chemical formulas are obligated to conform to a set of exacting criteria such as charge neutrality, balanced electronegativity, synthesizability, and mechanical stability. In response to this formidable task, we introduce a deep learning-based generative model for material composition and structure design by learning and exploiting explicit and implicit chemical knowledge. Our pipeline first uses deep diffusion language models as the generator of compositions and then applies a template-based crystal structure prediction algorithm to predict their corresponding structures, which is then followed by structure relaxation using a universal graph neural network-based potential. The density functional theory (DFT) calculations of the formation energies and energy-above-the-hull analysis are used to validate new structures generated through our pipeline. Based on the DFT calculation results, six new materials, including Ti2HfO5, TaNbP, YMoN2, TaReO4, HfTiO2, and HfMnO2, with formation energy less than zero have been found. Remarkably, among these, four materials, namely Ti2$HfO5, TaNbP, YMoN2, and TaReO4, exhibit an e-above-hull energy of less than 0.3 eV. These findings have proved the effectiveness of our approach.
Crystal structure prediction using neural network potential and age-fitness Pareto genetic algorithm
Sep 13, 2023While crystal structure prediction (CSP) remains a longstanding challenge, we introduce ParetoCSP, a novel algorithm for CSP, which combines a multi-objective genetic algorithm (MOGA) with a neural network inter-atomic potential (IAP) model to find energetically optimal crystal structures given chemical compositions. We enhance the NSGA-III algorithm by incorporating the genotypic age as an independent optimization criterion and employ the M3GNet universal IAP to guide the GA search. Compared to GN-OA, a state-of-the-art neural potential based CSP algorithm, ParetoCSP demonstrated significantly better predictive capabilities, outperforming by a factor of $2.562$ across $55$ diverse benchmark structures, as evaluated by seven performance metrics. Trajectory analysis of the traversed structures of all algorithms shows that ParetoCSP generated more valid structures than other algorithms, which helped guide the GA to search more effectively for the optimal structures
MD-HIT: Machine learning for materials property prediction with dataset redundancy control
Jul 10, 2023



Materials datasets are usually featured by the existence of many redundant (highly similar) materials due to the tinkering material design practice over the history of materials research. For example, the materials project database has many perovskite cubic structure materials similar to SrTiO$_3$. This sample redundancy within the dataset makes the random splitting of machine learning model evaluation to fail so that the ML models tend to achieve over-estimated predictive performance which is misleading for the materials science community. This issue is well known in the field of bioinformatics for protein function prediction, in which a redundancy reduction procedure (CD-Hit) is always applied to reduce the sample redundancy by ensuring no pair of samples has a sequence similarity greater than a given threshold. This paper surveys the overestimated ML performance in the literature for both composition based and structure based material property prediction. We then propose a material dataset redundancy reduction algorithm called MD-HIT and evaluate it with several composition and structure based distance threshold sfor reducing data set sample redundancy. We show that with this control, the predicted performance tends to better reflect their true prediction capability. Our MD-hit code can be freely accessed at https://github.com/usccolumbia/MD-HIT